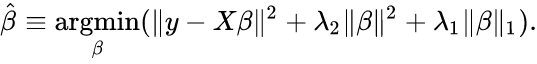$ cat file1.txt
1 apple
2 banana
3 pear
4 orange
$ cat file2.txt
1 pear x
2 apple y
3 banana z
5 kiwi q
$ join file1.txt file2.txt
1 apple pear x
2 banana apple y
3 pear banana z
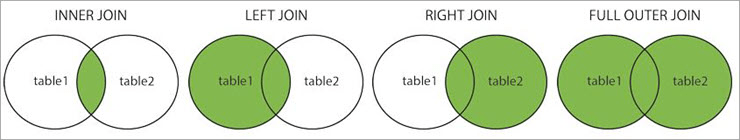
各种连接的概念示意图:
左/ 右 / 外连接 Left/right/outer join
-a N 输出指定第N个文件的所有行
通过-a选项可以完成left join,right join以及outer join的操作
单独指定-a 1 即为左连接(连接后输出第一个文件里所有的行)
单独指定 -a 2 即为右链接(连接后输出第二个文件里所有的行)
同时制定 -a 1 -a 2 为外连接(连接后输出两个文件里所有的行)
#left join
$ join -a 1 file1.txt file2.txt
1 apple pear x
2 banana apple y
3 pear banana z
4 orange
#right join
$ join -a 2 file1.txt file2.txt
1 apple pear x
2 banana apple y
3 pear banana z
5 kiwi q
#outer join
$ join -a 1 -a 2 file1.txt file2.txt
1 apple pear x
2 banana apple y
3 pear banana z
4 orange
5 kiwi q
Key
join默认使用的key是第一列,
-j N 指定两个文件j合并时所使用的key为第N列
也可以单独指定:
-1 N 指定第一个文件所用key为第N列
-2 N 指定第一个文件所用key为第N列
-e [字符串] 指定的key不存在时,输出“字符串”代替
#因为没有以第二列为key进行排序,所以报错
$ join -j 2 file1.txt file2.txt
join: file1.txt:4: is not sorted: 4 orange
join: file2.txt:2: is not sorted: 2 apple y
pear 3 1 x
#使用<(your command) 形式可以直接将 your command的结果作为输入
#使用排序后的结果则可以以第二列为key合并
join -j 2 <(sort -k 2 file1.txt) <(sort -k 2 file2.txt)
apple 1 2 y
banana 2 3 z
pear 3 1 x
#-j 2的效果等同 -1 2 -2 2
join -1 2 -2 2 <(sort -k 2 file1.txt) <(sort -k 2 file2.txt)
apple 1 2 y
banana 2 3 z
pear 3 1 x
#也可以使用-1 与-2 单独指定
#指定的key不存在时,输出“na”代替
$ join -j 4 -e na file1.txt file2.txt
na 1 apple 1 pear x
na 1 apple 2 apple y
na 1 apple 3 banana z
na 1 apple 5 kiwi q
na 2 banana 1 pear x
na 2 banana 2 apple y
na 2 banana 3 banana z
na 2 banana 5 kiwi q
na 3 pear 1 pear x
na 3 pear 2 apple y
na 3 pear 3 banana z
na 3 pear 5 kiwi q
na 4 orange 1 pear x
na 4 orange 2 apple y
na 4 orange 3 banana z
na 4 orange 5 kiwi q
-c --count 统计各行出现的次数
-u --unique 只输出不重复的行
-d --repeated 只输出重复的行
-D --all-repeated 输出全部重复的行
-i --ignore-case 比较时忽略大小写
-w N --check-chars=N 只比较行的前N个字符
-s N --skip-chars=N 比较时略过前N个字符,之比较N个字符后的内容
-f N --skip-fields=N 比较时略过前N个字段,之比较N个字段后的内容
以以下的文件为例
$ cat test_data.txt
green
red
white
blue
yellow
black
white
blue
blue
对没有sort排序的文件使用uniq,只删除连续重复的行(两个blue变为一个)
$ uniq test_data.txt
green
red
white
blue
yellow
black
white
blue
先sort后,再uniq即可全局删除重复 (删去了多余的blue和white)
$ sort test_data.txt | uniq
black
blue
green
red
white
yellow
使用-c可以统计行的重复次数
$ sort test_data.txt | uniq -c
1 black
3 blue
1 green
1 red
2 white
1 yellow
$ sort test_data.txt | uniq -c | sort
1 black
1 green
1 red
1 yellow
2 white
3 blue
使用-w可以只对前n个字符为对象进行删除 ,若n=1 , blue与black 则视为重复:
$ sort test_data.txt | uniq -w 1
black
green
red
white
yellow
$ cat test.txt
a 1
b 2
c 3
d 4
e 5
f 6
g 7
h 8
i 9
j 10
k 11
#head 查看开头,默认为十行
$ head test.txt
a 1
b 2
c 3
d 4
e 5
f 6
g 7
h 8
i 9
j 10
#tail 查看末尾,默认为十行
$ tail test.txt
b 2
c 3
d 4
e 5
f 6
g 7
h 8
i 9
j 10
k 11
head与 tail 使用 -n 选项可以 指定显示的行数
$ head -n 5 test.txt
a 1
b 2
c 3
d 4
e 5
tail -n 5 test.txt
g 7
h 8
i 9
j 10
k 11
-v verbose 输出时第一行显示文件名
head -v test.txt
==> test.txt <==
a 1
b 2
c 3
d 4
e 5
f 6
g 7
h 8
i 9
j 10
head , tail可以对多个文件使用
head test.txt test2.txt
==> test.txt <==
a 1
b 2
c 3
d 4
e 5
f 6
g 7
h 8
i 9
j 10
==> test2.txt <==
aaaaaa 123
aaaaab 234
aaabcd 345
使用-q (quiet)则不显示header
head -q test.txt test2.txt
a 1
b 2
c 3
d 4
e 5
f 6
g 7
h 8
i 9
j 10
aaaaaa 123
aaaaab 234
aaabcd 345
-b bytes
-c character
-f field
如果不指定的话会报错
cut test.txt
cut: you must specify a list of bytes, characters, or fields
Try 'cut --help' for more information.
单列提取
$cat test.txt
abc 1
bcd 2
cde 3
def 4
efg 5
#提取第二列(字节)
$ cut -c 2 test.txt
b
c
d
e
f
#提取第二列(字符)
$ cut -b 2 test.txt
b
c
d
e
f
#提取第二列(字段)
cut -f 2 -d ' ' test.txt
1
2
3
4
5
cut -b 1,3 test.txt
ac
bd
ce
df
eg
cut -b 1-3 test.txt
abc
bcd
cde
def
efg
cut -b 1-3,5 test.txt
abc1
bcd2
cde3
def4
efg5
$ cut -b -2 test.txt
ab
bc
cd
de
ef
cut -b 2- test.txt
bc 1
cd 2
de 3
ef 4
fg 5
多文件提取
cut可以同时对多个文件进行提取,输出时纵向连接
$ cat test.txt
abc 1
bcd 2
cde 3
def 4
efg 5
$ cat test2.txt
abc 123
bcd 234
cde 345
$cut -b 2 test.txt test2.txt
b
c
d
e
f
b
c
d
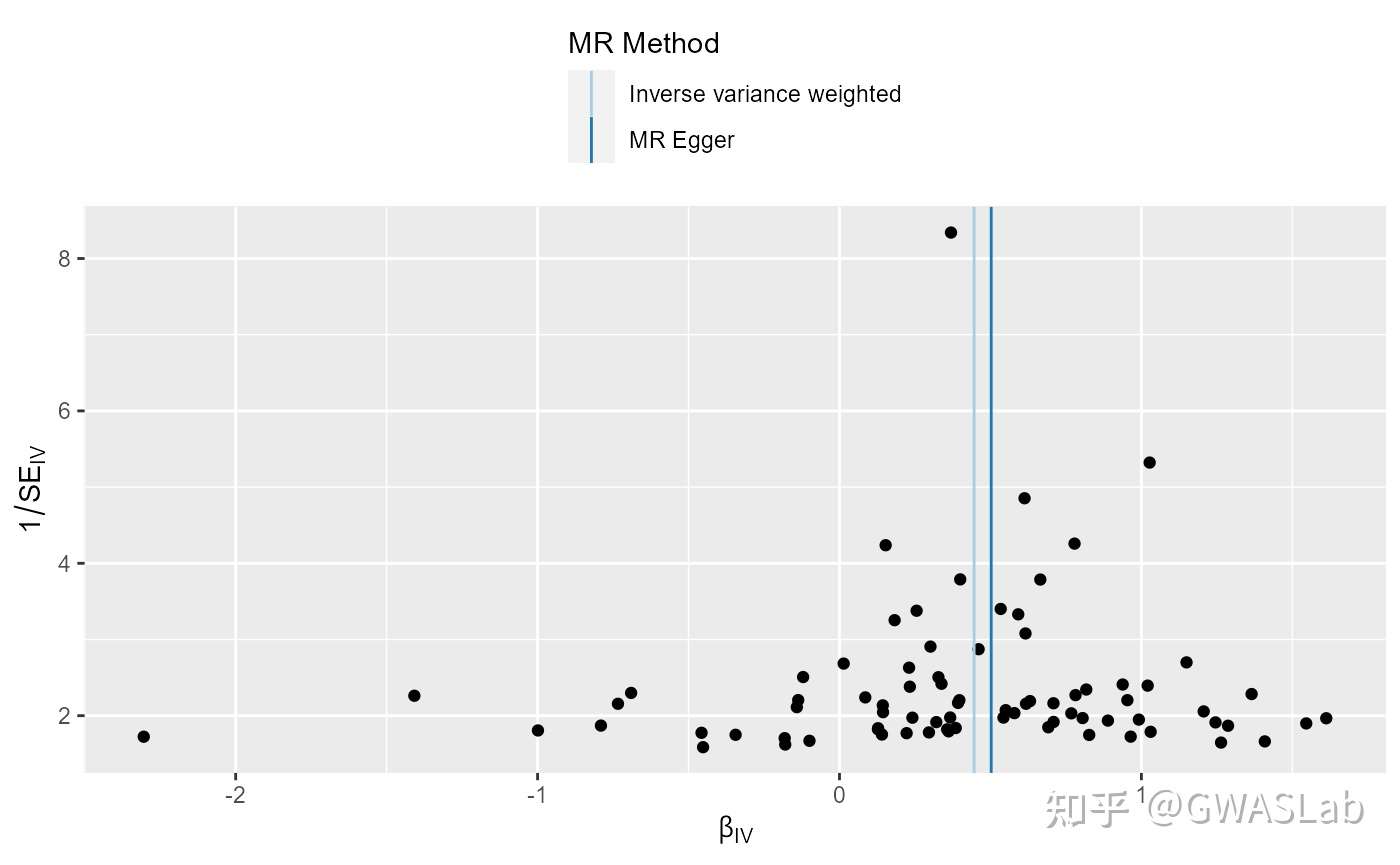
孟德尔随机化的原理可以参考G. Davey Smith and Ebrahim 2003; George Davey Smith and Hemani 2014, 统计方法可以参考Pierce and Burgess 2013; Bowden, Davey Smith, and Burgess 2015等。
bmi_exp_dat <- clump_data(bmi_exp_dat,clump_r2=0.01 ,pop = "EUR")
#> Please look at vignettes for options on running this locally if you need to run many instances of this command.
#> Clumping UVOe6Z, 30 variants, using EUR population reference
#> Removing 3 of 30 variants due to LD with other variants or absence from LD reference panel
ao <- available_outcomes()
#> API: public: <http://gwas-api.mrcieu.ac.uk/>
head(ao)
#> # A tibble: 6 x 22
#> id trait group_name year consortium author sex population unit
#> <chr> <chr> <chr> <int> <chr> <chr> <chr> <chr> <chr>
#> 1 ieu-b-~ Number o~ public 2021 MRC-IEU Clare~ Male~ European SD
#> 2 prot-a~ Leptin r~ public 2018 NA Sun BB Male~ European NA
#> 3 prot-a~ Adapter ~ public 2018 NA Sun BB Male~ European NA
#> 4 ukb-e-~ Impedanc~ public 2020 NA Pan-U~ Male~ Greater Midd~ NA
#> 5 prot-a~ Dual spe~ public 2018 NA Sun BB Male~ European NA
#> 6 eqtl-a~ ENSG0000~ public 2018 NA Vosa U Male~ European NA
#> # ... with 13 more variables: sample_size <int>, nsnp <int>, build <chr>,
#> # category <chr>, subcategory <chr>, ontology <chr>, note <chr>, mr <int>,
#> # pmid <int>, priority <int>, ncase <int>, ncontrol <int>, sd <dbl>
## 使用grep1抓取目标表型
ao[grepl("heart disease", ao$trait), ]
#> # A tibble: 28 x 22
#> id trait group_name year consortium author sex population unit
#> <chr> <chr> <chr> <int> <chr> <chr> <chr> <chr> <chr>
#> 1 ieu-a-8 Coronary h~ public 2011 CARDIoGRAM Schun~ Male~ European log ~
#> 2 finn-a~ Valvular h~ public 2020 NA NA Male~ European NA
#> 3 finn-a~ Other or i~ public 2020 NA NA Male~ European NA
#> 4 ukb-b-~ Diagnoses ~ public 2018 MRC-IEU Ben E~ Male~ European SD
#> 5 ukb-a-~ Diagnoses ~ public 2017 Neale Lab Neale Male~ European SD
#> 6 ukb-e-~ I25 Chroni~ public 2020 NA Pan-U~ Male~ South Asi~ NA
#> 7 ukb-b-~ Diagnoses ~ public 2018 MRC-IEU Ben E~ Male~ European SD
#> 8 finn-a~ Ischemic h~ public 2020 NA NA Male~ European NA
#> 9 finn-a~ Major coro~ public 2020 NA NA Male~ European NA
#> 10 finn-a~ Other hear~ public 2020 NA NA Male~ European NA
#> # ... with 18 more rows, and 13 more variables: sample_size <int>, nsnp <int>,
#> # build <chr>, category <chr>, subcategory <chr>, ontology <chr>, note <chr>,
#> # mr <int>, pmid <int>, priority <int>, ncase <int>, ncontrol <int>, sd <dbl>
Hemani G, Tilling K, Davey Smith G (2017) Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLOS Genetics 13(11): e1007081. https://doi.org/10.1371/journal.pgen.1007081
Hemani G, Zheng J, Elsworth B, Wade KH, Baird D, Haberland V, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Davey Smith G, Gaunt TR, Haycock PC, The MR-Base Collaboration. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018;7:e34408. doi: 10.7554/eLife.34408
Inouye, Michael, et al. “Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention.” Journal of the American College of Cardiology 72.16 (2018): 1883-1893.
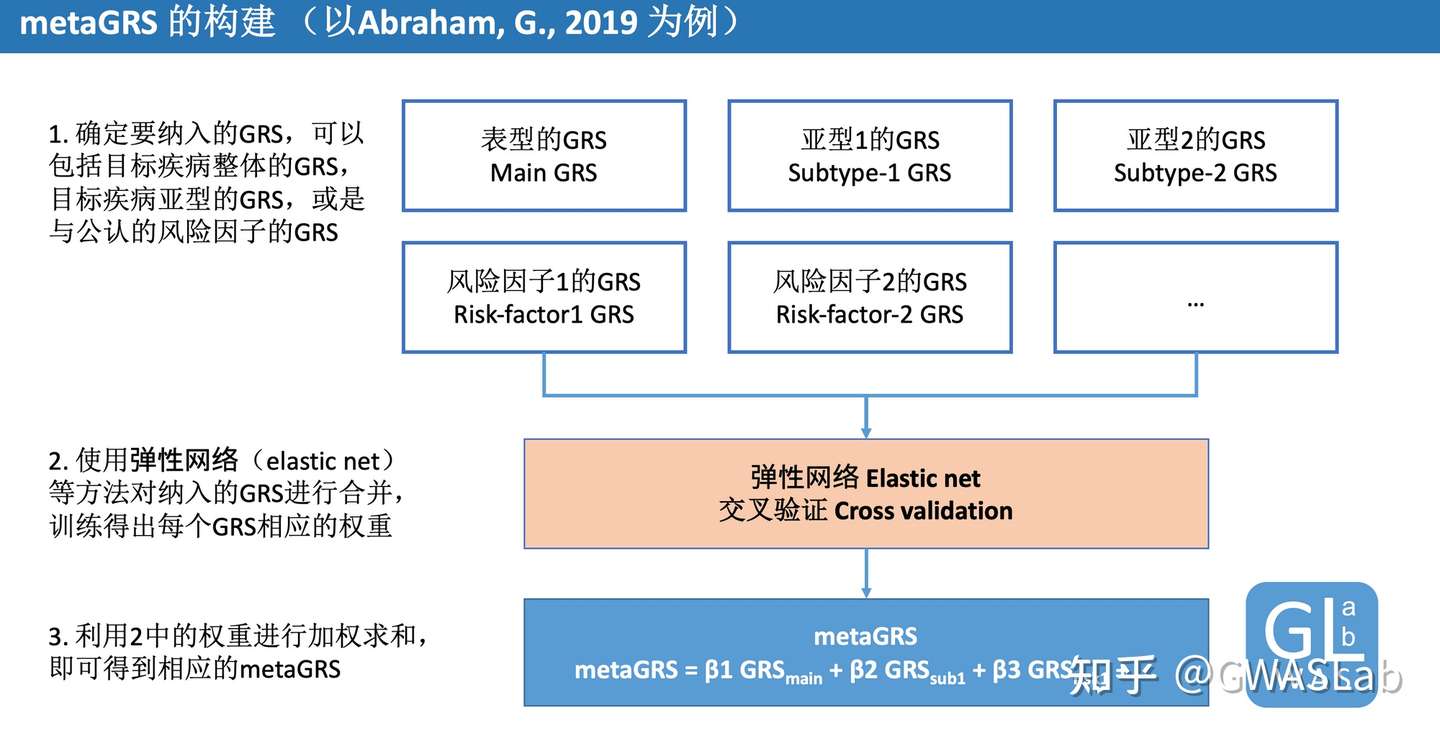
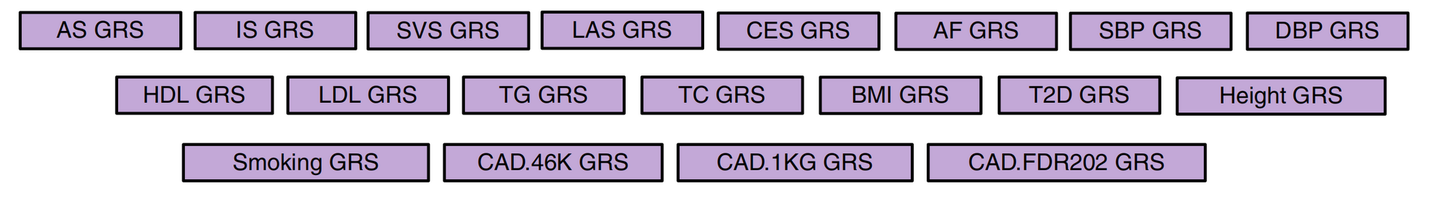
Abraham, G., Malik, R., Yonova-Doing, E. et al. Genomic risk score offers predictive performance comparable to clinical risk factors for ischaemic stroke. Nat Commun10, 5819 (2019). https://doi.org/10.1038/s41467-019-13848-1
Friedman, Jerome, Trevor Hastie, and Rob Tibshirani. “glmnet: Lasso and elastic-net regularized generalized linear models.” R package version 1.4 (2009): 1-24.