本文内容:
- MTAG简介
- 核心概念
- 核心假设
- 算法简介
- 特点与不足
- 使用教程
- 参考
MTAG简介
MTAG( multi-trait analysis of GWAS)是一个使用GWAS概括新数据进行多表型联合分析的方法,相比于单表型的GWAS,MTAG可以利用关联表型的信息提升目标表型的检验统计power。
核心概念
MTAG的核心概念是当某个表型的GWAS结果与其他表型的结果存在相关性时,就可以通过整合其他表性的效应量来提高该表型效应量的估计值。
核心假设
对于不同表型,所有的SNP都共享同一个效应量的方差协方差矩阵
MTAG算法简单介绍如下所示:

特点:
- 使用GWAS概括性数据,不需要个体基因型信息
- 所使用的GWAS概括性数据可以有样本重叠,不需要独立样本,因为MTAG会通过二变量LD分数回归来调整样本重叠可能带来的误差。(GWASLab:通过Bivariate LD Score regression估计遗传相关性 genetic correlation)
- MTAG可以计算对于特定表型每一个SNP的效应量估计值。
- MTAG计算相对高效,因为其每一步都有封闭解。
不足:
MTAG最主要的问题来源于那些与一个表型无关,而与另一个表型存在真实相关的SNP,MTAG对于此类SNP会有估计偏差,对于无关的SNP产生远离0的偏倚,造成假阳性。
使用教程
安装
MTAG是一个基于python的软件,可以从作者的github上下载:
https://github.com/JonJala/mtag
环境要求:
python2.7
numpy (>=1.13.1)scipypandas (>=0.18.1)argparsebitarray(forldsc)joblib
示例数据下载:
neuroticism 与 subjective well-being gwas的概括性数据:
http://ssgac.org/documents/1_OA2016_hm3samp_NEUR.txt.gz
http://ssgac.org/documents/1_OA2016_hm3samp_SWB.txt.gz
输入格式 (GWAS的概括性数据)
snpid chr bpos a1 a2 freq z pval n
snpid,chr,bpos分别为snp的rsID,染色体号,与碱基位置,
a1,a2分别为效应等位(effect allele),与非效应等位
freq为a1的频率
z,pval,n则分别为检验的Z分数,p值,与样本量
(也可以使用beta与se来代替z,-use_beta_se)
MTAG示例代码
python /[path]/mtag.py \
--sumstats 1_OA2016_hm3samp_NEUR.txt,1_OA2016_hm3samp_SWB.txt \
--out ./tutorial_results_1.1NS \
--n_min 0.0 \
--stream_stdout
sumstats: 输入的GWAS的概括性数据, 使用逗号间隔
out: 输出文件的前缀
n_min:样本量阈值 (n低于此阈值的SNP会被舍弃)
stream_stdout : 将log文件实时输出到终端
如果使用东亚的LD分数参考,还需要指定
-ld_ref_panel 这个选项所使用的数据与LDSC相同,可以通过LDSC下载 (GWASLab:连锁不平衡分数回归 LD score regression -LDSC)https://alkesgroup.broadinstitute.org/LDSCORE/
输出
总共四组文件,
.log文件_sigma_hat.txt存储估计的残差协方差矩阵_omega_hat.txt存储估计的遗传方差矩阵_trait_1.txt与_trait_2.txt文件则为MTAG的效应量估计结果
.log 文件:
head -n 20 tutorial_results_1.1NS.log
2021/10/31/10:43:32 PM
<><><<>><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><>
<>
<> MTAG: Multi-trait Analysis of GWAS
<> Version: 1.0.8
<> (C) 2017 Omeed Maghzian, Raymond Walters, and Patrick Turley
<> Harvard University Department of Economics / Broad Institute of MIT and Harvard
<> GNU General Public License v3
<><><<>><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><>
<> Note: It is recommended to run your own QC on the input before using this program.
<> Software-related correspondence: maghzian@nber.org
<> All other correspondence: paturley@broadinstitute.org
<><><<>><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><><>
Calling ./mtag.py \
--stream-stdout \
--n-min 0.0 \
--sumstats 1_OA2016_hm3samp_NEUR.txt,1_OA2016_hm3samp_SWB.txt \
--out ./tutorial_results_1.1NS
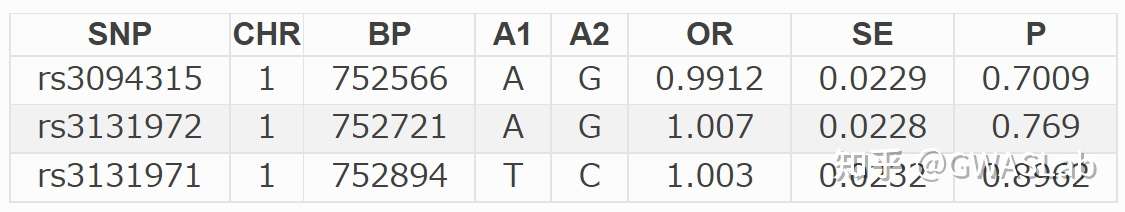
MTAG效应量估计值文件(trait_1):
head tutorial_results_1.1NS_trait_1.txt
SNP CHR BP A1 A2 Z N FRQ mtag_beta mtag_se mtag_z mtag_pval
rs2736372 8 11106041 T C -7.7161416126199995 111111.11111099999 0.4179 -0.032488048690724455 0.004191057650619415 -7.751754186899077 9.063170638233748e-15
rs2060465 8 11162609 T C 7.69444599845 62500.0 0.6194 0.038971244976047835 0.005364284755639267 7.264947099439278 3.731842884367329e-13
rs10096421 8 10831868 T G -7.561098219 111111.11111099999 0.4646 -0.0306226260844897350.004144573386350028 -7.388607518772383 1.483744201034853e-13
rs2409722 8 11039816 T G -7.382616018080001 111111.11111099999 0.4627 -0.032187004733709536 0.004145724613962613 -7.763903233057294 8.235474166666265e-15
rs11991118 8 10939273 T G 7.32202915636 111111.11111099999 0.5056 0.030603923980300953 0.004134432028802615 7.402207550419989 1.339389362466671e-13
rs2736371 8 11105529 A G -7.32009158327 62500.0 0.3806 -0.03759068256663257 0.005364284755639268 -7.007585219467501 2.42466338629309e-12
rs2736313 8 11086942 T C -7.24228035161 111111.11111099999 0.4646 -0.03068115211425502 0.004144573386350028 -7.402728641577938 1.3341420385130596e-13
rs876954 8 8310923 A G -7.15791677597 62500.0 0.4813 -0.03422779585744538 0.005212736369758894 -6.566185862767606 5.162037825451834e-11
rs1533059 8 8684953 A G -7.074128564239999 62500.0 0.4478 -0.035027431607855014 0.00523771146606734 -6.687545091932079 2.269452965596589e-11
其中mtag_beta mtag_se mtag_z mtag_pval列为这一个表型mtag计算后的结果。
参考:
Turley, P., Walters, R.K., Maghzian, O.et al.Multi-trait analysis of genome-wide association summary statistics using MTAG.Nat Genet50,229–237 (2018). https://doi.org/10.1038/s41588-017-0009-4