目录
- 公共数据库
- 各大Biobank与Cohort
- 欧美
- 东亚
- Global Biobank
- 单独的研究发表的数据
- 各类疾病研究组织的数据
本文将简要介绍可以简单入手的GWAS sumstats的一些来源,文中提及的来源都是公开可下载的,适合新入门的同学练手,做一些小规模的meta分析,或是post-gwas分析。
1.公共数据库:
目前收录最全的GWAS数据库,但只收录了已发表GWAS的数据,而且会有几个月到半年左右的延迟,使用时应该注意:
GWAS catalog: https://www.ebi.ac.uk/gwas/
OpenGWAS: https://gwas.mrcieu.ac.uk/
2.各大Biobank的公开数据:
规模较大的Biobank或Cohort会定期公开GWAS sumstats,这类数据有些未被收录在GWAS catalog中,一些gwas检验方法的作者也会使用其方法进行Biobank级别的GWAS分析,这类数据的表型覆盖较广:
2.1欧美:
Finngen 4 : https://r4.finngen.fi/about
Finngen 5 : https://r5.finngen.fi/about
Finngen 6 : https://r6.finngen.fi/about
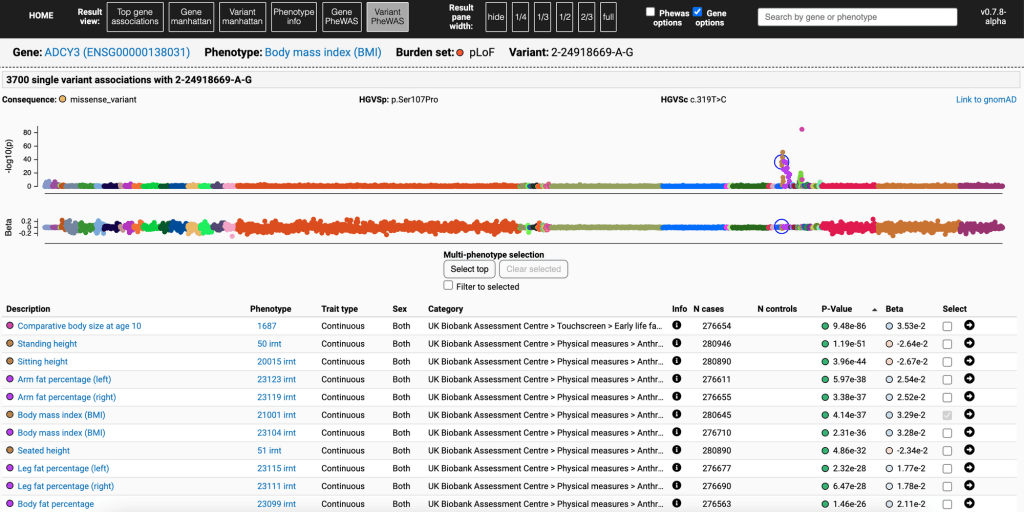
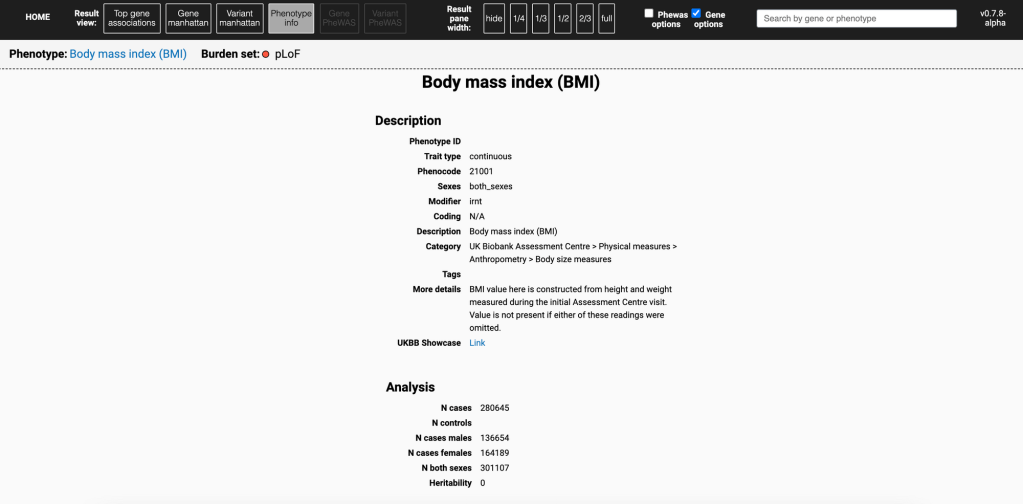
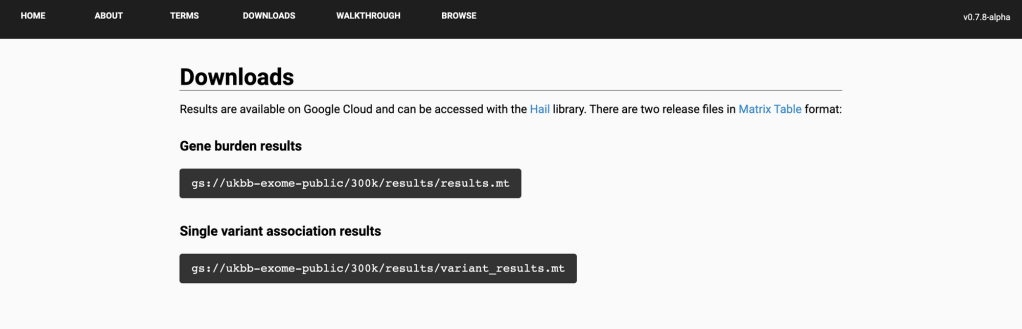
UKB : https://pheweb.org/UKB-Neale/
UKB saige: https://pheweb.org/UKB-SAIGE/
UKB fastgwa-glmm: https://yanglab.westlake.edu.cn/resources/ukb_fastgwa/imp_binary/
UKB fastgwa: https://yanglab.westlake.edu.cn/resources/ukb_fastgwa/imp/
UKB TOPMed: https://pheweb.org/UKB-TOPMed/
UKB gene-based: https://genebass.org/
Pan-UKB : https://pan.ukbb.broadinstitute.org/
MGI 1 : https://pheweb.org/MGI-freeze1/
MGI 2 : https://pheweb.org/MGI-freeze2/
MGI BioUV: https://pheweb.org/MGI-BioVU/
FinMetSeq: https://pheweb.sph.umich.edu/FinMetSeq/
Generation Scotland: https://datashare.ed.ac.uk/handle/10283/844
2.2 东亚:
Biobank Japan:
JENGER: http://jenger.riken.jp/result
Pheweb: https://pheweb.jp/
ToMMo – JMorp:https://jmorp.megabank.tohoku.ac.jp/202109/gwas/
KoreanChip: https://www.koreanchip.org/downloads
KoGES Pheweb: https://koges.leelabsg.org/
2.3 全球范围Biobank的Meta分析:
近期Global Biobank项目也公开了一批全球范围biobank的常见复杂疾病meta分析
Global Biobank :http://results.globalbiobankmeta.org/
3.单独的研究发表的数据
直接在google上搜索研究论文,从Data availability里的url顺藤摸瓜找到数据,此类数据散落在各个学校,研究机构等等自家的网站上,没有好的办法,只有自己搜索。GWAS catalog 经常会出现收录不及时而漏掉很多最新数据。
例如:
Program in Complex Trait Genomics, IMB, The University of Queensland.
https://cnsgenomics.com/content/data
https://ctg.cncr.nl/software/summary_statistics
4.各类疾病研究组织的数据
这类数据通常为meta分析后的sumstats,例如:
DIAGRAM:http://www.diagram-consortium.org/downloads.html
Megastroke: https://www.megastroke.org/index.html
GIANT (Genetic Investigation of ANthropometric Traits):https://portals.broadinstitute.org/collaboration/giant/index.php/Main_Page
GLGC (Global Lipids Genetics Consortium): http://csg.sph.umich.edu/willer/public/glgc-lipids2021/
PGC (Psychiatric Genomics Consortium): https://www.med.unc.edu/pgc/download-results/
等等
一个人的总结难免会有疏漏,欢迎大家在评论里补充!