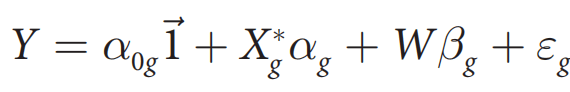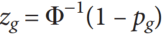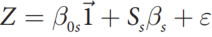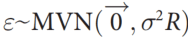基因组学与单细胞RNA测序的结合
目前复杂疾病研究方法热点之一就是多组学多方法的结合,近来多种新的结合方法中,scDRS是比较有代表性的将基因组学与单细胞RNA测序的结合的方法,类似的方法还有sc-linker等,这类方法也属于一个新的交叉领域单细胞遗传学(Single-cell Genetics)。
传统的基因组学与单细胞测序的结合的方法,例如MAGMA或是LDSC-SEG本质上还是基于细胞特异表达的基因集而进行的富集检测,而近来以scDRS为代表的方法则利用scRNA-seq的表达矩阵进一步深入至单个细胞的层面,能够或得更高的分辨率,这对解析疾病异质性,疾病关联的细胞亚群等方面可以发挥巨大作用 (就是让精准医疗更精准)。
scDRS的全称是 single-cell disease relevance score, 单细胞疾病相关分数,正如其名称类似于多基因分风险分数的PRS,该方法结构上或多或少也类似PRS的构建和计算方法,不过这里的个体是一个一个的细胞。PRS用于评估个体疾病的风险,而scDRS则评估细胞是否高表达疾病相关的基因。注意这里的R,一个是risk风险 (有方向),一个是relevance相关(没有方向),概念上的差异。
scDRS的方法概况

- 首先从GWAS结果构建疾病基因集:使用MAGMA和目标疾病的GWAS sumstats进行基因水平的关联检验,得到每个基因与疾病关联的Z分数,选定前1000个基因作为假定的疾病基因 (这个步骤不是方法的重点,除了MAGMA也可以使用其他类似方法构建疾病基因集)
- 然后计算单个细胞的疾病分数:scDRS会对每个细胞进行计算,量化假定疾病基因的整体表达。为了最大化检验效能,会根据MAGMA所得Z分数进行加权,同时根据每个基因在单细胞测序中特异的技术性噪音进行进行加权。
- 最后scDRS对所有基因集和细胞,标准化其原始的疾病分数以及原始的对照分数。然后基于所有基因集和细胞的标准化后分数的经验分布,计算每个细胞的P值。
- 利用得到的分数可以进行多种下游分析,分析包括(1)单个细胞层面的关联性检验(2)细胞类型关联性检验 以及(3)基因关联性检验等, 这些分析的P值均通过MC检验(Monte Carlo 蒙特卡洛检验)获得。
用例(下游分析)
分析与表型相关的细胞类型
相比于传统方法LDSC或MAGMA,scDRS可以检验细胞类型的相关性,还可以检验同一细胞类型内的异质性。
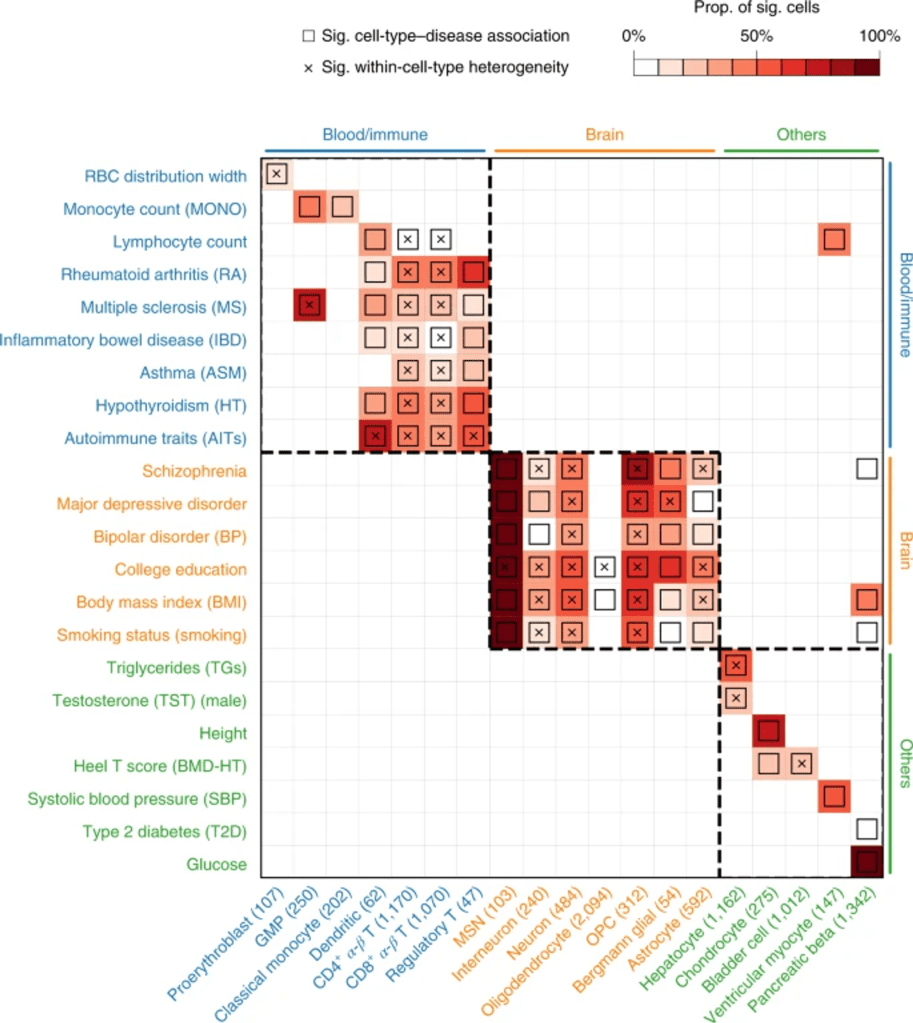
作者列举了 22个表型与19种细胞的关联结果的热力图,Y轴为表型,X轴为细胞类型,方框表示显著相关,×表示细胞类型内的异质性,颜色深浅表示显著关联细胞的比例。
发现存在异质性的细胞亚群
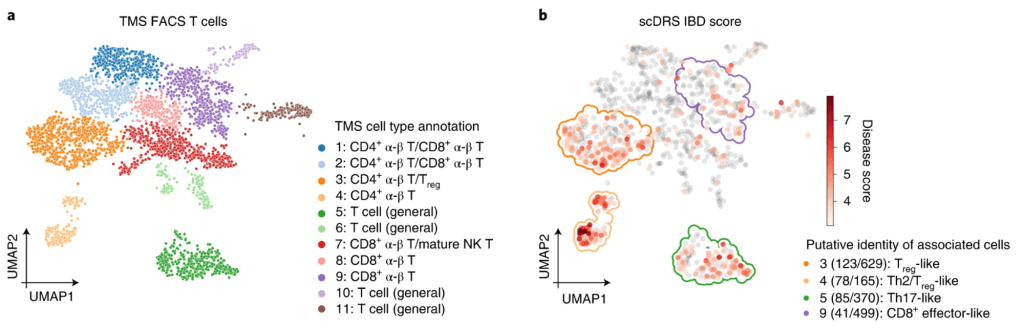
作者研究了自身免疫性疾病中T细胞的异质性,11个T细胞群中(a),与IBD相关的T细胞构成了4个新的群(b)。
基因-分数的关联分析
scDRS检验基因是否与GWAS由来的基因集中的基因共表达。
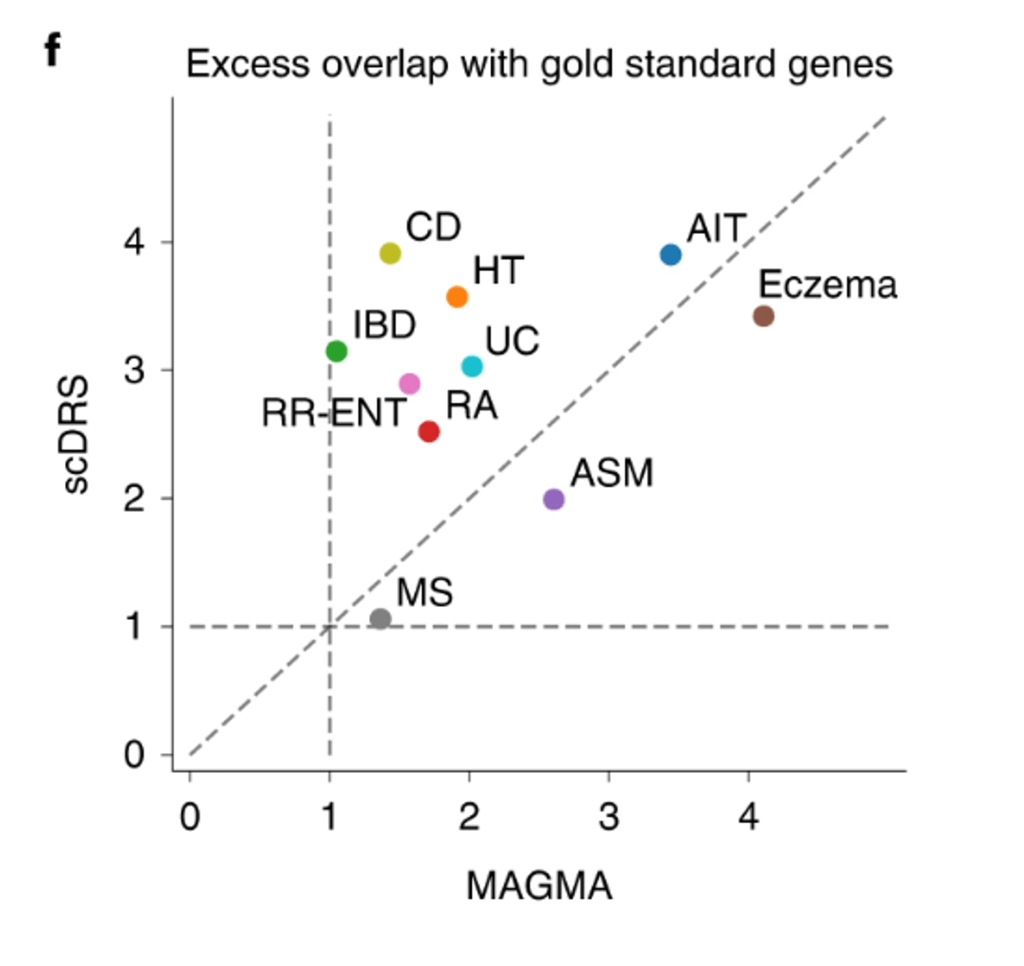
相比于MAGMA,scDRS能更准确地识别出疾病相关的基因
典型的分析流程
只需要GWAS的Sumstats和单细胞RNA测序的数据,好消息是这两个都可以很容易从公开数据库中获得。
具体流程官方文档以及大牛博客已经写得很详细了,
分析代码可以参考 https://martinjzhang.github.io/scDRS/
以及 https://zhuanlan.zhihu.com/p/592128325
注意的点
- scDRS的假设是基因集里的基因与疾病有关,与疾病有关的基因会在疾病相关的细胞群体(可以是疾病细胞或健康细胞)里高表达,与基因的方向无关。 scDRS并不是假设基因集里的基因会在疾病细胞里高表达。注意这里概念上细节的差别。(https://github.com/martinjzhang/scDRS/issues/42)
- 基本数据要求:为了能够得到足够的检验效能,GWAS的heritability z-score最好大于5,或样本量大于10万。(https://martinjzhang.github.io/scDRS/faq.html#which-gwas-and-scrna-seq-data-to-use)
- Seurat格式的数据需要转换为scanpy使用的h5ad格式(表达矩阵不能有负值,Seurat的scaled.data里表达矩阵会有负值,转换时要注意)(https://github.com/martinjzhang/scDRS/issues/44);转换可以使用 SeuratDisk https://mojaveazure.github.io/seurat-disk/articles/convert-anndata.html
- 为了增加检验效能,单细胞RNA测序可以事先进行 imputation (https://github.com/martinjzhang/scDRS/issues/32)
- -adj-prop 可以调整细胞类别的比例 (当某些细胞种类比例过高的时候) (https://github.com/martinjzhang/scDRS/issues/32)
- 显著细胞的数太少:正常现象,仍然可以进行group分析(检验效能通常更高) (https://martinjzhang.github.io/scDRS/faq.html#scdrs-detected-few-significant-cells-fdr-0-2)
参考
Zhang, M. J., Hou, K., Dey, K. K., Sakaue, S., Jagadeesh, K. A., Weinand, K., … & Price, A. L. (2022). Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data. Nature genetics, 54(10), 1572-1580.