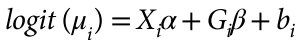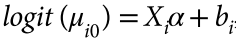Marees, A.T., de Kluiver, H., Stringer, S., Vorspan, F., Curis, E., Marie-Claire, C., and Derks, E.M. (2018). A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. International Journal of Methods in Psychiatric Research 27.
FID1 Family ID for first sample
IID1 Individual ID for first sample
FID2 Family ID for second sample
IID2 Individual ID for second sample
RT Relationship type inferred from .fam/.ped file
EZ IBD sharing expected value, based on just .fam/.ped relationship
Z0 P(IBD=0)
Z1 P(IBD=1)
Z2 P(IBD=2)
PI_HAT Proportion IBD, i.e. P(IBD=2) + 0.5*P(IBD=1)
PHE Pairwise phenotypic code (1, 0, -1 = AA, AU, and UU pairs, respectively)
DST IBS distance, i.e. (IBS2 + 0.5*IBS1) / (IBS0 + IBS1 + IBS2)
PPC IBS binomial test
RATIO HETHET : IBS0 SNP ratio (expected value 2)