
GWAS研究中发现的显著SNP只能解释人类群体中复杂性状很小一部分的遗传变异。那么剩余的遗传力在哪里? 很多时候这部分遗传力并没有丢失,而是由于部分snp效应太小以至于无法达到显著水平而没有被检测到。
与单SNP关联检验相对,GCTA中的GREML(genome-based restricted maximum likelihood)方法使用线性混合模型( linear mixed models , LMMs),将全部SNP的效应作为随机效应拟合。
模型如下:

y是 一个n × 1 的表型向量,n是样本大小; β 是固定效应的向量,诸如性别、年龄、以及一个或多个主成分(PC); u 是SNP效应的向量,服从如下分布:
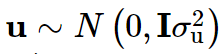
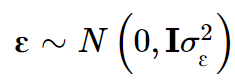
W是标准化的基因型矩阵,其中第ijth 个元素为:
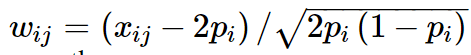
其中 xij 是第j个个体的第i个SNP的参考allele的拷贝数, pi该 allele 的频率。如果我们定义 A = WW′/N 并且定义 σ2g 为全部SNP解释的方差 , 即 σ2g=Nσ2u, (N为SNP的数量),那么方程 1 等价于:
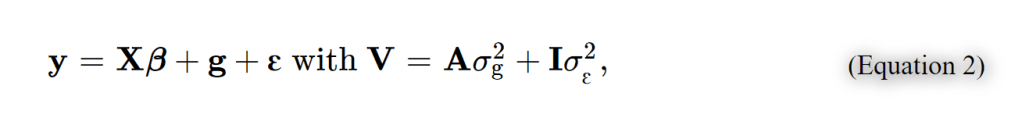
其中g是一个n x 1的向量,表示各个个体的全部的遗传效应,服从 g∼N(0,Aσ2g) 。A 可以理解为个体两两间遗传关系组成的遗传关系矩阵 genetic relationship matrix (GRM) 。于是基于这个线性混合效应模型我们就可以通过估计的GRM,利用REML( restricted maximum likelihood )算法来无偏地估计全部SNP解释的方差 σ2g (继而估计SNP遗传力)。
基于以上定义,个体j与个体k的遗传关联可以由以下方程估计:
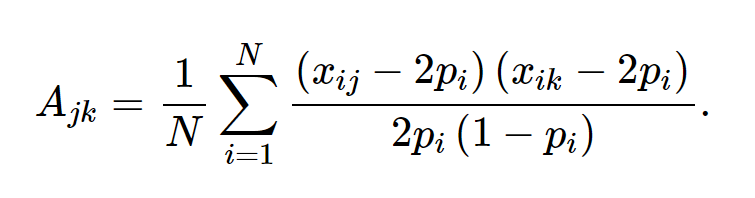
实际操作中我们首先需要排除掉有亲缘关系的个体,主要原因是该模型的目的是估计所有SNP解释的方差,如果纳入有亲缘关系的个体,会由于表型关联( phenotypic correlations )而造成偏差,例如由于共同的环境因素。即使没有上述的偏差,那么对它的解读也会有别于 “无关联的”个体:一个基于家系的估计值捕捉的是所有因果变异的贡献,而该方法捕捉的则是与被基因分型的SNP处于LD的因果变异的贡献。
下面简单介绍如何利用GCTA软件估计snp-遗传力:
GCTA下载链接:https://cnsgenomics.com/software/gcta/#Download
GCTA语法上类似PLINK,文件格式也几乎通用,使用起来十分便捷:
第一步: 估计GRM
gcta64 --bfile test --autosome --maf 0.01 --make-grm --out test --thread-num 10
输入为plink格式的bed/bim/fam文件,
- –autosome 只是用常染色体上的SNP
- –maf 0.01 过滤掉maf小于0.01的snp
- –make-grm 计算GRM
- –out 输出文件的前缀
- –thread-num 10 要使用的线程数
计算完成后GRM会存储在以下文件中: test.grm.bin, test.grm.N.bin and test.grm.id
我们也可以去除掉有亲缘关系的个体:
gcta64 --grm test --grm-cutoff 0.025 --make-grm --out test_rm025
第二步:利用GCTA-GREML估计SNP遗传力
gcta64 --grm test --pheno test.phen --reml --out test --thread-num 10
- –pheno 表型文件
- –reml 利用reml算法计算snp遗传力
计算结果储存在 test.hsq的文件中
当然我们以可以加入协变量,例如前10个主成分 PC1-10:
gcta64 --grm test --pheno test.phen --reml --qcovar test_10PCs.txt --out test --thread-num 10
对于较大的数据量,我们还可以分染色体进行计算,详见:https://cnsgenomics.com/software/gcta/#Tutorial
参考:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3014363/
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nature Genetics 42, 565–569 (2010).

One thought on “使用GCTA (GREML)来估计SNP-遗传力 SNP Heritability”