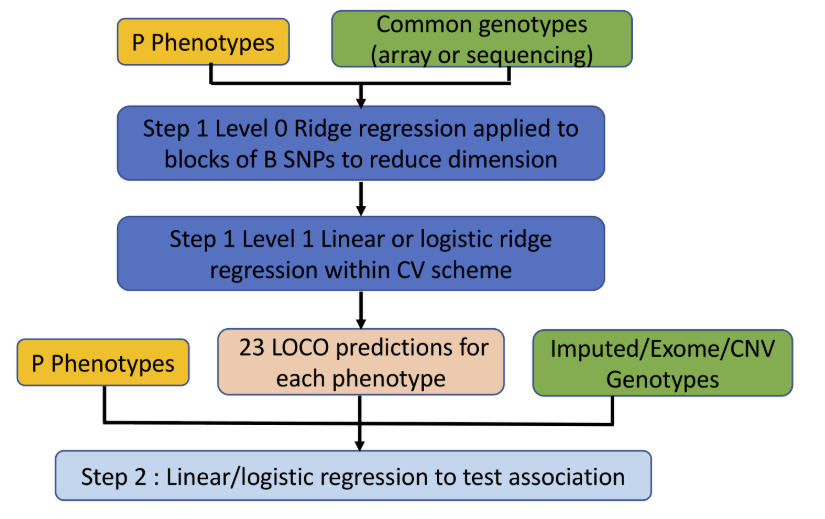
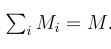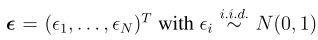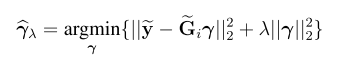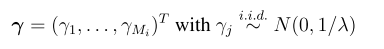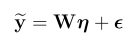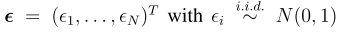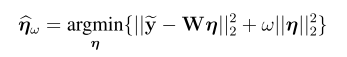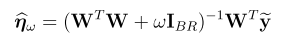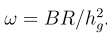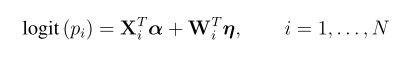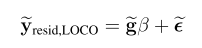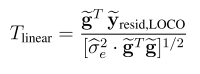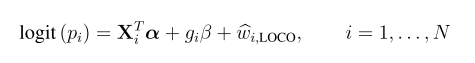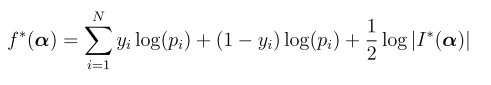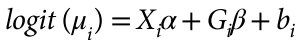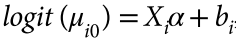head pgc.cross.SCZ17.2013-05.txt
snpid hg18chr bp a1 a2 or se pval info ngt CEUaf
rs3131972 1 742584 A G 1 0.0966 0.9991 0.702 0 0.16055
rs3131969 1 744045 A G 1 0.0925 0.9974 0.938 0 0.133028
rs3131967 1 744197 T C 1.001 0.0991 0.9928 0.866 0 .
rs1048488 1 750775 T C 0.9999 0.0966 0.9991 0.702 0 0.836449
rs12562034 1 758311 A G 1.025 0.0843 0.7716 0.988 0 0.0925926
rs4040617 1 769185 A G 0.9993 0.092 0.994 0.979 0 0.87156
rs4970383 1 828418 A C 1.096 0.1664 0.5806 0.439 0 0.201835
rs4475691 1 836671 T C 1.059 0.1181 0.6257 1.02 0 0.146789
rs1806509 1 843817 A C 0.9462 0.1539 0.7193 0.383 0 0.600917
Metadata:
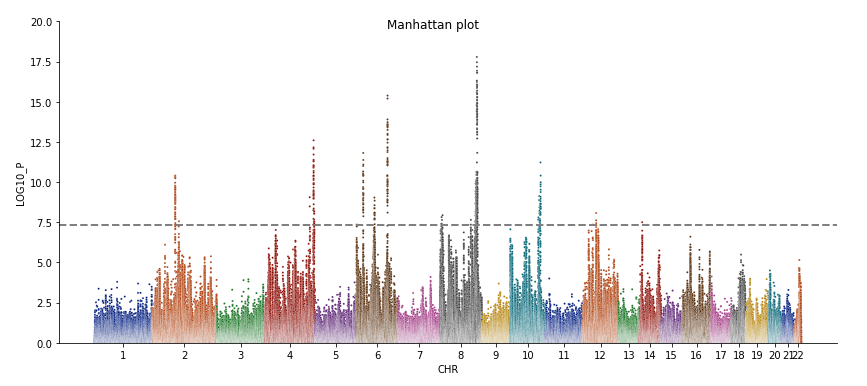
Mean chi^2 = 1.229
Lambda GC = 1.201
Max chi^2 = 32.4
11 Genome-wide significant SNPs (some may have been removed by filtering).
Conversion finished at Mon Apr 4 13:21:29 2016
Total time elapsed: 16.07s
Bulik-Sullivan, B., Loh, PR., Finucane, H. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet47, 291–295 (2015). https://doi.org/10.1038/ng.3211
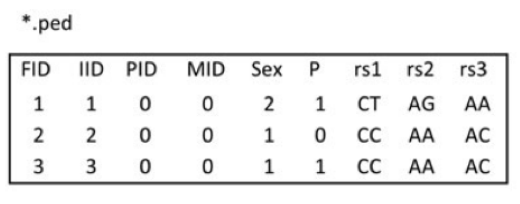
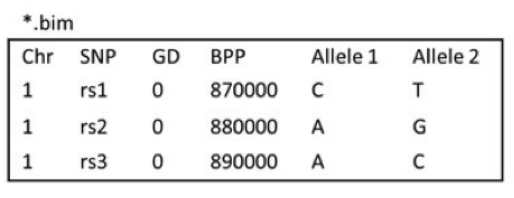
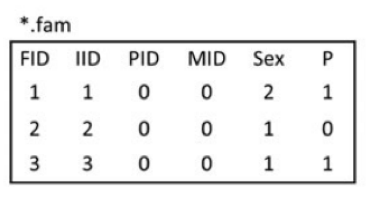
1.ped,pedigree file。ped文件一般包含6+2N列,第一至六列分别为 1. Family ID 2. Individual ID 3.Mother ID 4.Father ID 5.Sex 6.Phenotype。第六列以后为各个SNP的等位基因,两列一组,可以使用具体的碱基,也可以使用拷贝数(0,1)。
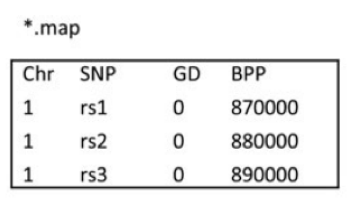
2. map,与ped文件相伴随的文件,主要包含ped文件中SNP的位置信息。一般包含4列。分别是1. 染色体号 2.SNP ID 3.遗传图距(单位为摩根或厘摩,通常分析不需要这一列,使用哑值(dummy value) 0 填充) 4.碱基对坐标。每行一个SNP,顺序与ped文件中的SNP相对应。
downloaded data (VCF.gz and tab-indices), ~ 15.5 GB
converted BCF files and their indices, ~14 GB
binary PLINK files, ~53 GB
pruned PLINK binary files, ~ <1 Gb
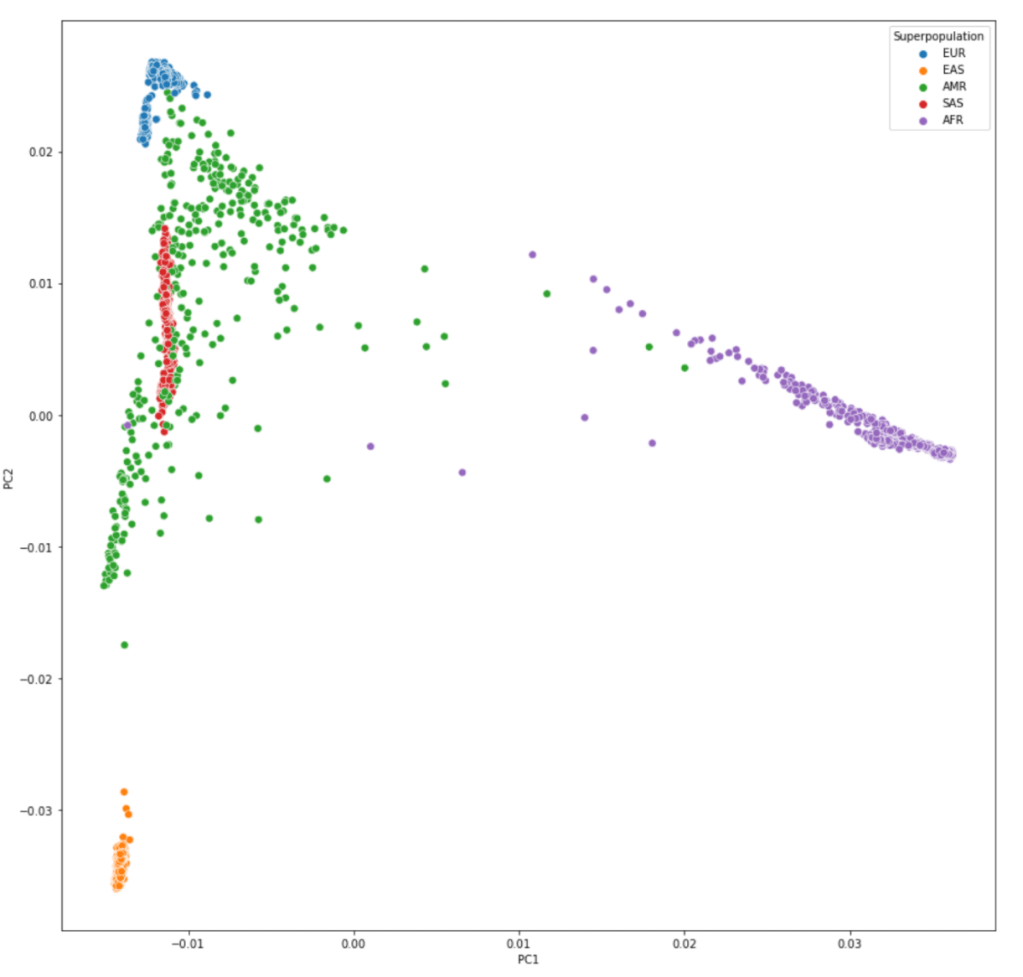
2.2 下载所用数据 (本教程数据主要来自1000 Genomes Phase III imputed genotypes)
2.3 下载对应每条染色体的vcf文件
prefix="ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ALL.chr" ;
suffix=".phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf.gz" ;
for chr in {1..22}; do
wget "${prefix}""${chr}""${suffix}" "${prefix}""${chr}""${suffix}".tbi ;
done
2.4 下载 1000 Genomes Phase III 的 ped文件,包含了个体性别,群体等基本信息
#Populations
This file describes the population codes where assigned to samples collected for the 1000 Genomes project. These codes are used to organise the files in the data_collections' project data directories and can also be found in column 11 of many sequence index files.
There are also two tsv files, which contain the population codes and descriptions for both the sub and super populations that were used in phase 3 of the 1000 Genomes Project:
<ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/20131219.populations.tsv>
<ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/20131219.superpopulations.tsv>
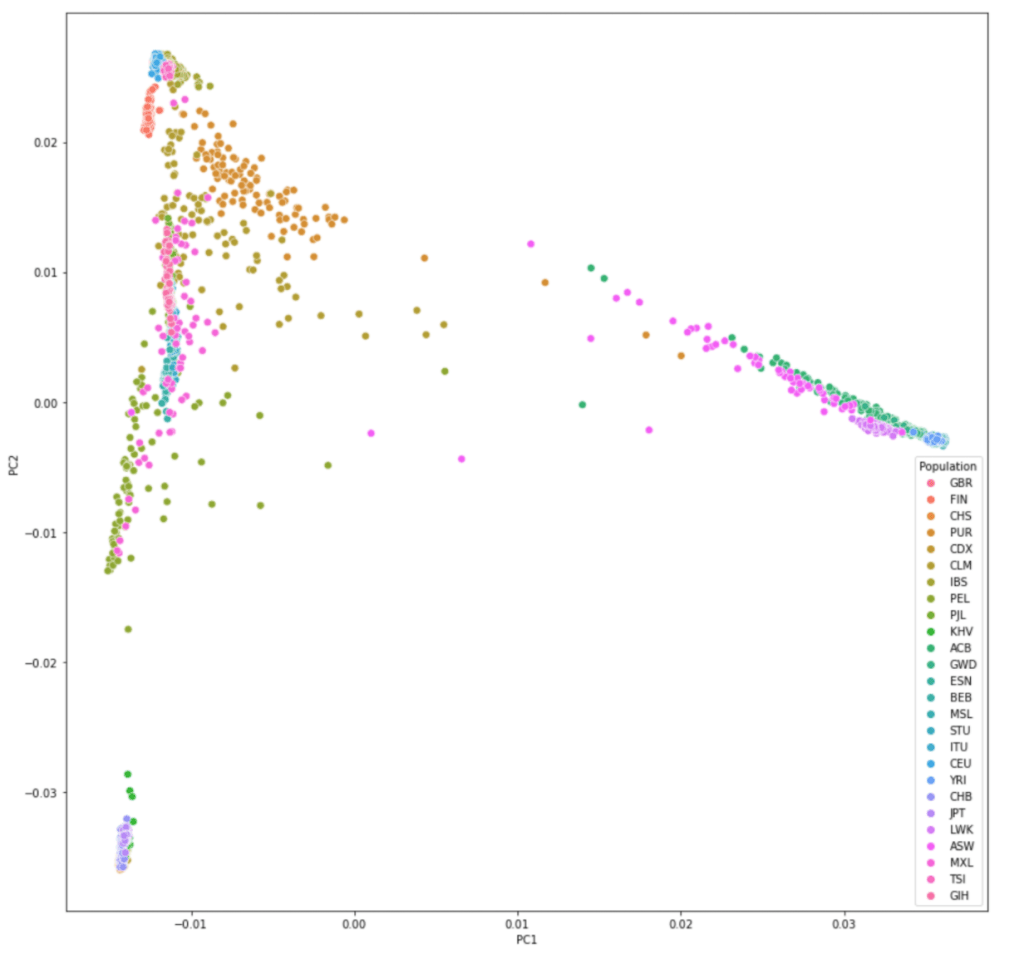
###Populations and codes
CHB Han Chinese Han Chinese in Beijing, China
JPT Japanese Japanese in Tokyo, Japan
CHS Southern Han Chinese Han Chinese South
CDX Dai Chinese Chinese Dai in Xishuangbanna, China
KHV Kinh Vietnamese Kinh in Ho Chi Minh City, Vietnam
CHD Denver Chinese Chinese in Denver, Colorado (pilot 3 only)
CEU CEPH Utah residents (CEPH) with Northern and Western European ancestry
TSI Tuscan Toscani in Italia
GBR British British in England and Scotland
FIN Finnish Finnish in Finland
IBS Spanish Iberian populations in Spain
YRI Yoruba Yoruba in Ibadan, Nigeria
LWK Luhya Luhya in Webuye, Kenya
GWD Gambian Gambian in Western Division, The Gambia
MSL Mende Mende in Sierra Leone
ESN Esan Esan in Nigeria
ASW African-American SW African Ancestry in Southwest US
ACB African-Caribbean African Caribbean in Barbados
MXL Mexican-American Mexican Ancestry in Los Angeles, California
PUR Puerto Rican Puerto Rican in Puerto Rico
CLM Colombian Colombian in Medellin, Colombia
PEL Peruvian Peruvian in Lima, Peru
GIH Gujarati Gujarati Indian in Houston, TX
PJL Punjabi Punjabi in Lahore, Pakistan
BEB Bengali Bengali in Bangladesh
STU Sri Lankan Sri Lankan Tamil in the UK
ITU Indian Indian Telugu in the UK
Should you have any queries, please contact info@1000genomes.org.
下面演示用PYTHON进行绘图:
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
Marees, A.T., de Kluiver, H., Stringer, S., Vorspan, F., Curis, E., Marie-Claire, C., and Derks, E.M. (2018). A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. International Journal of Methods in Psychiatric Research 27.
FID1 Family ID for first sample
IID1 Individual ID for first sample
FID2 Family ID for second sample
IID2 Individual ID for second sample
RT Relationship type inferred from .fam/.ped file
EZ IBD sharing expected value, based on just .fam/.ped relationship
Z0 P(IBD=0)
Z1 P(IBD=1)
Z2 P(IBD=2)
PI_HAT Proportion IBD, i.e. P(IBD=2) + 0.5*P(IBD=1)
PHE Pairwise phenotypic code (1, 0, -1 = AA, AU, and UU pairs, respectively)
DST IBS distance, i.e. (IBS2 + 0.5*IBS1) / (IBS0 + IBS1 + IBS2)
PPC IBS binomial test
RATIO HETHET : IBS0 SNP ratio (expected value 2)

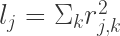
![E[\chi^2|l_j] = {{Nh^2l_j}\over{M}} + Na + 1](https://s0.wp.com/latex.php?latex=E%5B%5Cchi%5E2%7Cl_j%5D+%3D+%7B%7BNh%5E2l_j%7D%5Cover%7BM%7D%7D+%2B+Na+%2B+1&bg=ffffff&fg=333333&s=3&c=20201002)