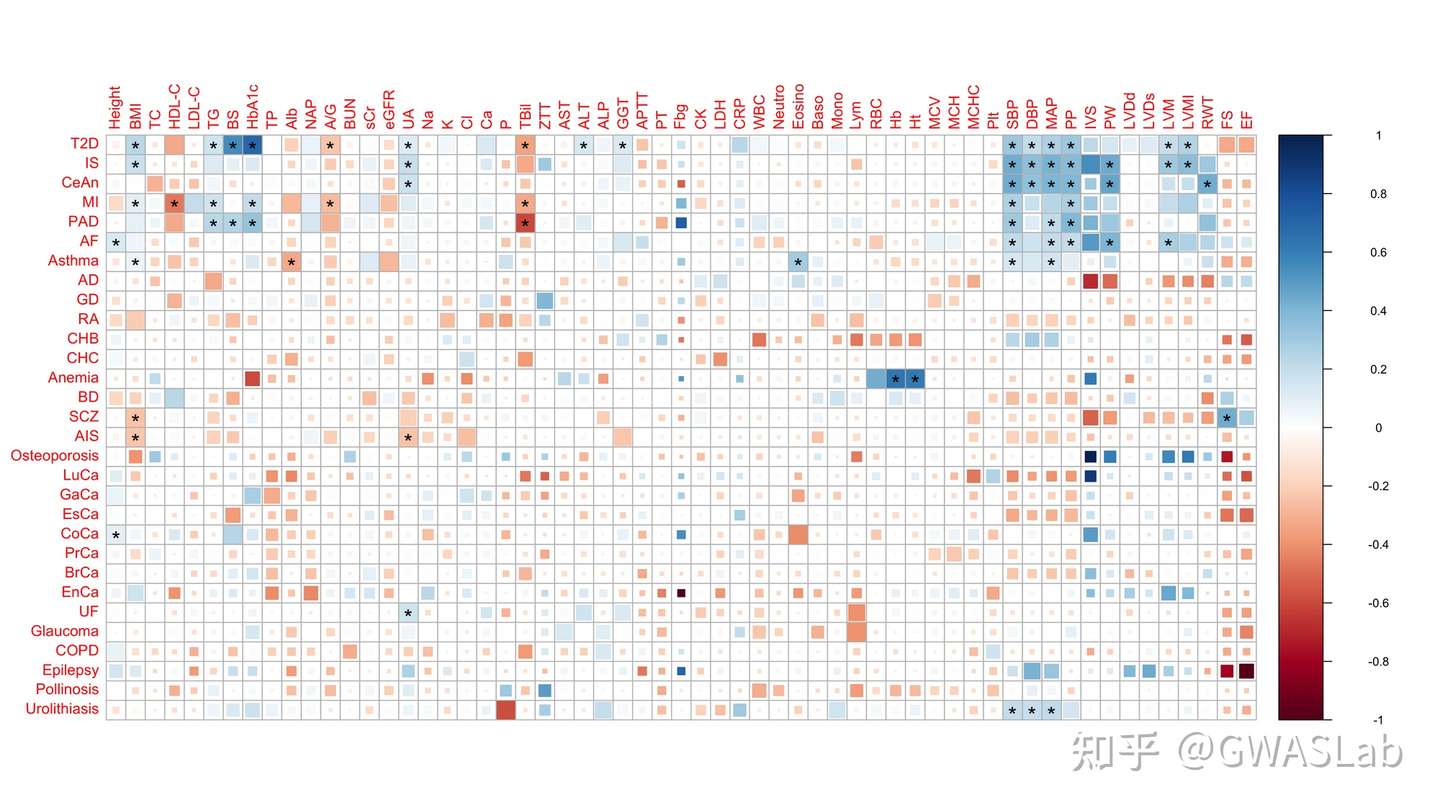
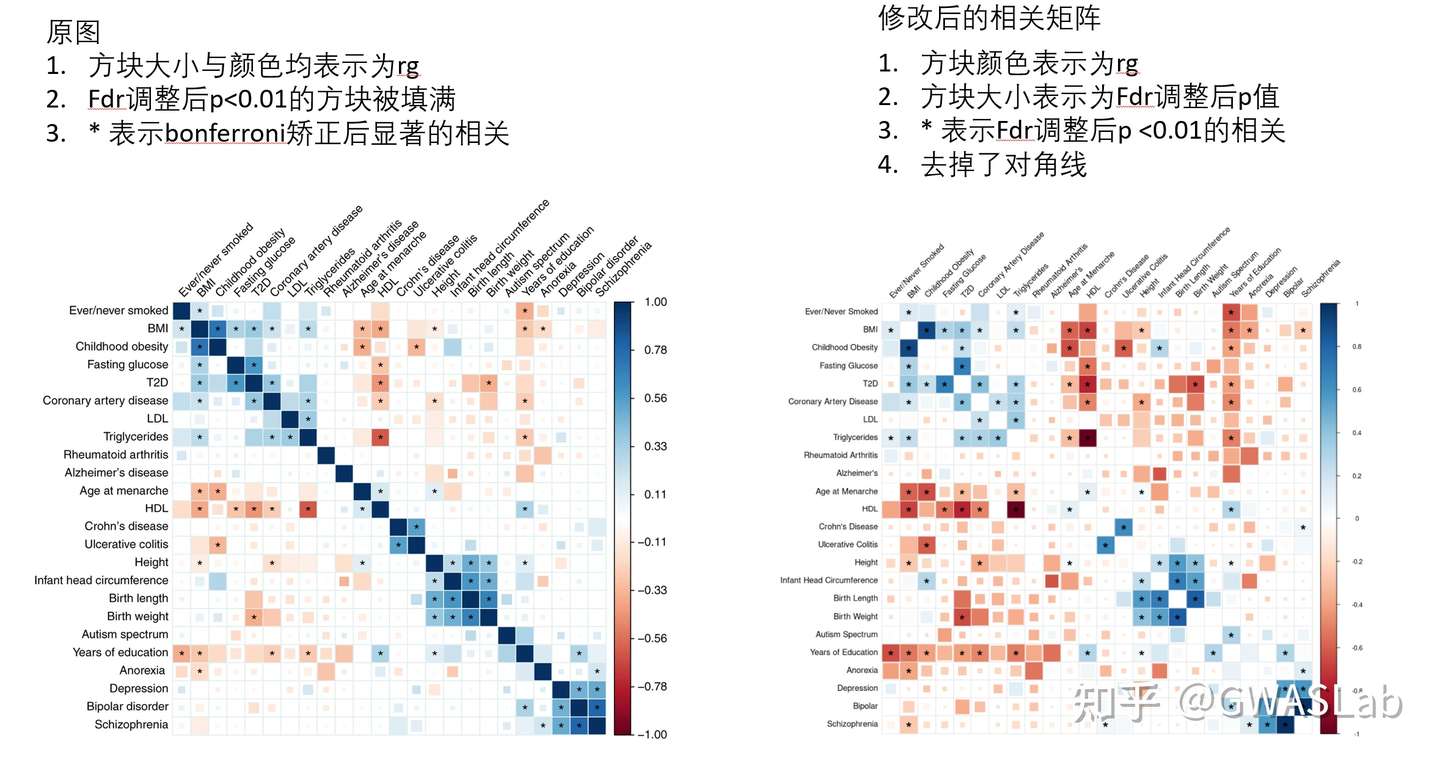
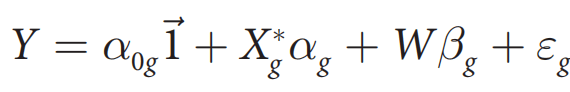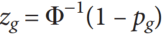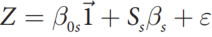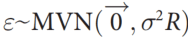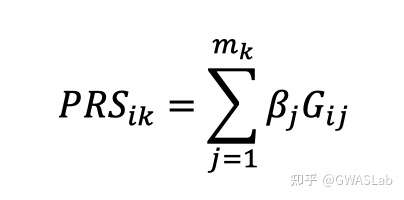popcorn 背景介绍
回顾(在单一族裔中估计遗传相关的方法):通过Bivariate LD Score regression估计遗传相关性 genetic correlation
随着GWAS等遗传变异关联研究在不同人群中不断开展,相关研究数量不断增加,这为我们提供了宝贵的机会以研究遗传结构在不同人种中的差异,有助于解决这个在医疗研究与群体遗传学中十分重要的问题。
该文章基于Brielin等的研究,简单介绍一种估计跨族裔遗传相关的方法,popcorn,用于估计在不同群体中常见SNP的因果效应量的相关性。该方法纳入了研究中所有SNP的关联信息(区别于其他需要事先进行LD-pruning的方法),而且仅使用GWAS的概括数据,减少了计算负担,并避免了无法获取基因型数据的问题。
通常情况下,在单一群体中,两个表性之间的遗传相关的定义为对于两个表型的SNP效应量大小之间的相关系数。而在多群体的情况时,因为不同群体内等位基因频率有显著差异,定义并不统一。因为某个变异可能在某个特定群体中有较高的效应量但较低的频率,所以本方法不仅考虑了效应量大小的相关,同时也考虑遗传冲击(allelic impact)的相关性。
该文中,跨族裔遗传效应相关的定义为SNP效应量的相关系数,跨族裔遗传冲击相关的定义为特定群体的等位方差正态化(allele-variance-normalized)的SNP效应量之间的相关系数。
直觉上,遗传效应相关性(genetic-effect correlation)衡量的是同一个SNP造成表型改变的程度差异,而遗传冲击相关性(genetic-impact correlation)则是分别在各个群体中,相比于的稀有allele,给予common allele更高的权重。举个例子,如果只看效应相关性,计算得出的相关系数是不完全的,没有反应出两个群体间的效应的完整信息,因为即使效应量的遗传相关是1,但其在两个群体中,对其各自表型分布的冲击也可能由于allele frequency的不同而不同。
所以跨族裔遗传相关的两个核心问题就被引出:
1 同一个变异在两个群体中效应量大小的差异?
2 同一个变异的效应量在两个群体中的差异是否被不同群体的allele frequency所稀释或加重?
为了估计遗传相关,popcorn采用了贝叶斯方法,假设基因型是分别从各个群体内抽取的,并且效应量服从正态的先验分布(无穷小模型)。无穷小模型的假设会产生一个观测统计量(Z scores)的多元正态分布,其中该分布的协方差是遗传力与遗传相关的一个方程。这使得我们可以直接将Z score的膨胀纳入模型中,而不需要进行LD pruning。然后popcorn会最大化近似加权似然方程,来估计遗传力与遗传相关性。
方程推导可参考原文章,这里我们要估计的是以下两个值:
遗传效应相关:pge=Cor(β1,β2)
遗传冲击相关:pgi=Cor(δ1β1,δ2β2)
其中δ为allele的标准差
使用方法:
该文章作者的github仓库: https://github.com/brielin/Popcorn
要求:
popcorn只能使用python2,建议用conda新建一个环境
需要以下python包:
numpy 1.14.2
scipy 1.0.1
pandas 0.22.0
pysnptools 0.3.9
bottleneck 1.0.0
statsmodels 0.8.0
(to use --plot_likelihood) matplotlib 1.5.1
从github clone之后,安装:
cd Popcorn
python setup.py install
可以下载使用千人基因组项目提前计算好的LD分数 (用于EUR-EAS):
https://www.dropbox.com/sh/37n7drt7q4sjrzn/AAAa1HFeeRAE5M3YWG9Ac2Bta .
popcorn 方法分为两步,
第一步为计算LD分数(使用下载好的数据可以跳过这一步,作者提供了EUR-EAS的LD分数)
第二步为计算遗传相关性
第一步,计算LD分数(可选)
python -m Popcorn compute -v 2 --bfile1 Popcorn/test/EUR_ref --bfile2 Popcorn/test/EAS_ref --gen_effect Popcorn/test/EUR_EAS_test_ge.cscore
-v 表示 输出log的详细程度
—bfile表示 输入的plink bfile文件前缀
—gen_effec 表示计算遗传效应相关 (计算遗传冲击相关时不加这个flag)
最后是输出的路径和文件名
第一步log文件
Popcorn version 0.9.6
(C) 2015-2016 Brielin C Brown
University of California, Berkeley
GNU General Public License v3
Invoking command: python /home/brielin/CoHeritability/Popcorn/__main__.py compute -v 2 --bfile1 Popcorn/test/EUR_ref --bfile2 Popcorn/test/EAS_ref Popcorn/test/EUR_EAS_test.cscore
Beginning analysis at DATE
50000 SNPs in file 1
50000 SNPs in file 2
49855 SNPs in file 1 after MAF filter
44021 SNPs in file 2 after MAF filter
43898 SNPs common in both populations
T1
T2
TC
Analysis finished at DATE
结果的格式如下所示:
1 14930 rs75454623 A G 0.520874751491 0.58630952381 1.4252539451 1.78489131664 1.49214493091
1 15211 rs78601809 T G 0.731610337972 0.503968253968 1.20243142615 2.3876817626 1.29403313474
1 15820 rs200482301 T G 0.728628230616 0.605158730159 1.56556914093 1.42934198599 1.40055911225
1 55545 rs28396308 T C 0.813121272366 0.804563492063 2.59950434496 2.1809181648 2.06610287872
各列分别代表Chromosome, Base Position, SNP ID, Allele 1, Allele 2, Frequency of A1 in POP1, Frequency of A1 in POP2, LD score in POP1, LD score in POP2, and Cross-covariance score
第二步,计算相关
python -m Popcorn fit -v 1 --use_mle --cfile Popcorn/test/EUR_EAS_test_ge.cscore --gen_effect --sfile1 Popcorn/test/EUR_test.txt --sfile2 Popcorn/test/EAS_test.txt Popcorn/test/EAS_EUR_test_ge_result.txt
–sfile1与–sfile2为输入的两个群体的GWAS文件,注意要与LD分数中POP1和POP2的顺序相一致
输入的GWAS数据至少包含一下列(列名应与下面的介绍保持一致):
1 rsid 或者 SNP (与LD分数文件中的id保持一致)
注: 可以使用 chr 以及 pos 来替代
2 a1 和 a2
3 N 每个SNP的样本大小
4 beta和SE 或者 OR和p-value 或者 Z分数 (不要求对应a1为effect allele,但要保持所有snp一致即可)
5 AF (可选)即a2的allele frequency,用于A/T , G/C的SNP的flip
输入文件例如:
rsid a1 a2 af N beta SE
rs10458597 C A 0.59421 50000 -0.0152782557183 0.00912151085515
rs3131972 A C 0.83102 50000 -0.000638379272685 0.0119663325893
rs3131969 A C 0.8654 50000 -0.000737906302463 0.0131325861467
rs3131967 A C 0.87044 50000 -0.000189441590945 0.0133506462369
执行代码后的,第二步log文件:
Popcorn version 0.9.6
(C) 2015-2016 Brielin C Brown
University of California, Berkeley
GNU General Public License v3
Invoking command: python /home/brielin/CoHeritability/Popcorn/__main__.py fit -v 1 --cfile Popcorn/test/EUR_EAS_test_ge.cscore --gen_effect --sfile1 Popcorn/test/EUR_test.txt --sfile2 Popcorn/test/EAS_test.txt Popcorn/test/EAS_EUR_test_ge_result.txt
Beginning analysis at DATE
Analyzing a pair of traits in two populations
49999 49999 SNPs detected in input.
43897 SNPs remaining after merging with score file.
43897 SNPs remaining after filtering on AF and self-compliment SNPs.
Initial estimate:
Val (obs) SE
h1^2 0.172133 NaN
h2^2 0.086708 NaN
pg 0.302372 NaN
Performing jackknife estimation of the standard error using 200 blocks of size 219 .
Jackknife iter: 200
Val (obs) SE Z P (Z)
h1^2 0.172133 0.012870 13.374388 0
h2^2 0.086708 0.008555 10.135759 0
pge 0.302372 0.053021 13.157553 0
Analysis finished at DATE
Total time elapsed: T
pge 则为跨族裔遗传相关的估计值 (加–gen_effect时的结果)
pgi 为跨族裔遗传冲击的估计值(不加–gen_effect时的结果)
这里的p值的原假设是估计值等于1,p<0.05则意味着估计值显著小于1
参考: