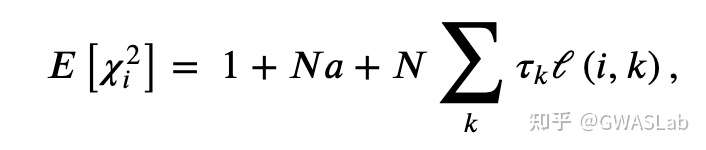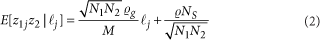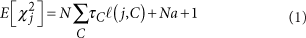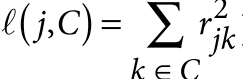目录
基于PLINK文件计算LD分数
LD分数文件格式
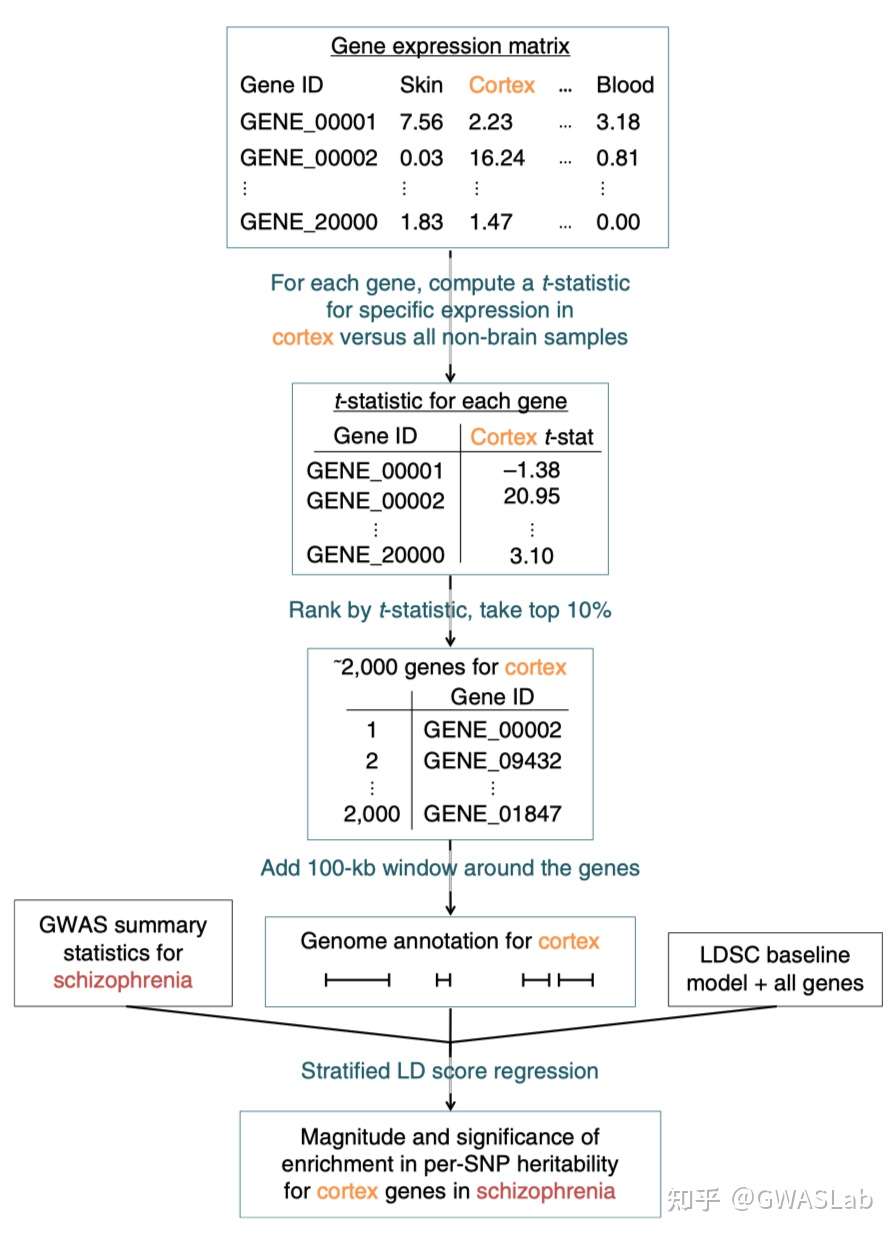
用于LDSC-SEG的分层LD分数的计算
有很多时候没有现成的LD分数参考,而需要我们自己根据手头基因型数据而计算LD分数(例如一个常用的例子是GWAS数据与scRNA数据的结合 – 使用自己的scRNA数据提取特征基因后计算LD分数并进行partitioned LD 分数回归以检测细胞特异性)。
本文将简要介绍如何计算 单变量的LD分数 以及 分层的LD分数。
回顾:
- GWASLab:连锁不平衡分数回归 LD score regression -LDSC
- GWASLab:通过Bivariate LD Score regression估计遗传相关性 genetic correlation
- GWASLab:基于功能分类分割遗传力 – 分层LD分数回归 Stratified LD score regression
- GWASLab:使用GWAS数据进行组织细胞特异性分析 – LDSC-SEG
使用LDSC计算单变量LD分数
单变量LD分数为最基本的LD分数的形式,常用在ldsc -h2选项的参考LD分数中。
输入文件: PLINK的 bed / bim / fam文件
使用软件: ldsc
这里推荐使用千人基因组相应族裔的数据(vcf格式),可通过plink -vcf转化为plink格式。
计算LD分数时需要指定窗口的大小,作者推荐 1 centiMorgan (cM) window
一般情况下plink格式中 表示cM的列都为零,可以使用plink —cm-map以及额外的遗传图谱文件来填补bim文件中的cM列
(从ldsc网站下载的plink文件中bim文件的cM列已经被填补,可以直接使用)
下面简单演示如何计算:
示例数据为千人基因组欧洲人群的第22号染色体:
wget <https://data.broadinstitute.org/alkesgroup/LDSCORE/1kg_eur.tar.bz2>
tar -jxvf 1kg_eur.tar.bz2
22.bim
22.fam
22.bed
22.hm3.daf.gz
可以通过以下的代码来计算ld分数 (-l2 选项)
python ldsc.py \
--bfile ${plinkfile} \ #指定plink文件路径及前缀
--l2 \ #计算ld分数
--ld-wind-cm 1 \ #指定窗口的大小,单位为cM
--out 22 #输出文件的前缀
执行上述代码后得到如下四个文件:
22.log
22.l2.M
22.l2.M_5_50
22.l2.ldscore.gz
LD分数相关文件格式
下面简要介绍LD分数文件的相关格式,主要的输出包括以下五种文件:
.log
.l2.ldscore
.annot
.l2.M_5_50
.l2.M
.log LD分数计算后得到的日志文件,其中会包含LD分数的总结性数据,例如:
Summary of LD Scores in 22.l2.ldscore.gz
MAF L2
mean 0.232 18.535
std 0.145 16.104
min 0.001 0.066
25% 0.104 7.839
50% 0.224 13.484
75% 0.355 22.972
max 0.500 109.716
MAF/LD Score Correlation Matrix
MAF L2
MAF 1.000 0.275
L2 0.275 1.000
Analysis finished at Fri Jan 30 10:58:46 2015
Total time elapsed: 2.72s
存储LD分数最主要的文件:
.l2.ldscore 文件
包含3+N列,N≥1, 前3列分别为
- CHR — chromosome 染色体号
- SNP — SNP identifier (rs number) rsID
- BP — physical position (base pairs) 物理位置
第三列以后为LD分数的列,可以有多列,对应不同的注释组
- all additional columns — LD Scores
CHR SNP BP L2
22 rs9617528 16061016 1.271
22 rs4911642 16504399 1.805
22 rs140378 16877135 3.849
22 rs131560 16877230 3.769
22 rs7287144 16886873 7.226
22 rs5748616 16888900 7.379
22 rs5748662 16892858 7.195
22 rs5994034 16894090 2.898
22 rs4010554 16894264 6.975
.annot注释文件
上面单变量LD分数的计算中我们使用的是plink文件里所有的SNP,未使用到注释文件,但后续分层LD分数计算时,我们需要用到annot文件,来注释计算LD分数时使用的SNP
该文件每行对应一个SNP,空格分隔,有表头,需要按以下顺序排列:
- CHR — chromosome 染色体号
- BP — physical position (base pairs) 物理位置
- SNP — SNP identifier (rs number) rsID
- CM — genetic position (centimorgans) 遗传距离
- all additional columns — Annotations 一列或多列注释
注释列既可以为0/1二分类,也可是连续的。
例如 Example:
CHR BP SNP CM AN1 AN2
1 1 rs1 0 1 0
1 2 rs2 0 1 0
1 3 rs3 0 0 1
最后则是两个辅助文件:
.l2.M_5_50
该文件只有一行,列数对应着.l2.ldscore文件中annotation中列的个数,并与其顺序一致。
每一列的数字则表示对应的注释分类中MAF>5%的SNP的个数。
(没有附加注释文件时就是该染色体上全部MAF>5%的SNP数量)
.l2.M
与l2.M_5_50相同,只是没有MAF的限制,即表示对应注释总的SNP数
例如:(对应两个annotation分类)
100 1000
计算分层LD分数
通过某种方式,选取注释组中包含的基因
例如使用单细胞测序中某个细胞特异表达的前10%的基因等,
注意:目前ldsc只支持常染色体。
为了计算某种注释特有的LD分数,需要准备一个.annot的注释文件,格式如上所述。
这个注释里的位点的顺序应该与.bim文件中的SNP完全相同。ldsc也允许—thin-annot 格式,也就是上述annot文件忽略掉CHR,BP,SNP以及CM列。
下面介绍如何构建annot文件并以此计算相应注释的LD分数:
Step 1: Creating an annot file 第一步,构建注释文件
输入文件:
—gene-set-file:注释包含的基因集
head GTEx_Cortex.GeneSet
ENSG00000105605
ENSG00000169862
ENSG00000074317
ENSG00000196361
ENSG00000221946
–gene-coord-file:基因坐标文件
head ENSG_coord.txt
GENE CHR START END
ENSG00000243485 chr1 29554 31109
ENSG00000237613 chr1 34554 36081
ENSG00000186092 chr1 69091 70008
ENSG00000238009 chr1 89295 133566
ENSG00000239945 chr1 89551 91105
–windowsize:基因上下游LD计算时所包含窗口大小
–bimfile:计算LD所用PLINK文件中的bim文件
head 1000G.EUR.QC.22.bim
22 rs587616822 -0.0048996974 16050840 G C
22 rs62224609 -0.00094708154 16051249 C T
22 rs587646183 0.010833912 16052463 C T
22 rs139918843 0.012979739 16052684 C A
22 rs587743102 0.014465964 16052837 T C
–annot-file:输出文件
所使用的代码如下所示:使用lsmake_annot.py
python make_annot.py \
--gene-set-file GTEx_Cortex.GeneSet \
--gene-coord-file ENSG_coord.txt \
--windowsize 100000 \
--bimfile 1000G.EUR.QC.22.bim \
--annot-file GTEx_Cortex.annot.gz
也可以直接使用bed文件计算
python make_annot.py \
--bed-file Brain_DPC_H3K27ac.bed \
--bimfile 1000G.EUR.QC.22.bim \
--annot-file Brain_DPC_H3K27ac.annot.gz
结果如下:
zcat GTEx_Cortex.annot.gz | head
ANNOT
0
0
0
0
0
Step 2: Computing LD scores with an annot file. 第二步,使用注释文件计算LD分数
使用第一步计算所得的.annot文件(忽略掉了CHR,BP,SNP以及CM列,所以使用–thin-annot选项)
–print-snps hm.22.snp:只打印包含在hm.22.snp的SNP
python ldsc.py \
--l2 \
--bfile 1000G.EUR.QC.22 \
--ld-wind-cm 1\
--annot Brain_DPC_H3K27ac.annot.gz \
--thin-annot \
--out Brain_DPC_H3K27ac\
--print-snps hm.22.snp
如果要与baseline model一起使用的话,要注意使用的SNP应该完全相同。
正式计算时只需要对所有染色体循环即可,确保输出都在同一个文件夹里,且有着相同的前缀,例如:
${prefix}.${chr}.annot.gz , ${prefix}.${chr}.l2.ldscore , and ${prefix}.${chr}.l2.M_5_50
这些文件可以用于后续分层LDSC回归分析中。
GWASLab:使用GWAS数据进行组织细胞特异性分析 – LDSC-SEG