
目录
- TwoSampleMR简介
- TwoSampleMR简要教程
- 方法概览
- 读取暴露GWAS数据,选取工具变量
- 读取结局GWAS数据,提取对应工具变量
- 对数据进行预处理,使其效应等位与效应量保持统一
- MR分析与可视化
- MR分析
- 敏感性分析
- 散点图
- 森林图
- 漏斗图
- 参考
TwoSampleMR简介
两样本孟德尔随机化(Two sample Mendelian randomisation,2SMR)是使用GWAS概括性数据估计暴露对结果的因果效应的一种方法。尽管孟德尔随机化在概念上相对直观简单,但实际操作却相对繁琐,有很多细节需要注意。TwoSampleMR就是一款致力于简化MR分析的R包,其整合了以下的分析步骤:
- 数据管理与统一
- 因果推断的常规统计学分析
- 接入了GWAS概括性数据的数据库,使分析更为便捷
孟德尔随机化的原理可以参考G. Davey Smith and Ebrahim 2003; George Davey Smith and Hemani 2014, 统计方法可以参考Pierce and Burgess 2013; Bowden, Davey Smith, and Burgess 2015等。
TwoSampleMR简要教程
本文仅对TwoSampleMR使用方法进行介绍。基础概念概念可以参考:GWASLab:孟德尔随机化系列之一:基础概念 Mendelian randomization I
下载(注:本文所用语言均为R)
TwoSampleMR官网: https://mrcieu.github.io/TwoSampleMR/
TwoSampleMR是一个R包,可以通过以下代码在R中直接安装
install.packages("remotes")
remotes::install_github("MRCIEU/TwoSampleMR")
0 方法概览
MR分析的主要步骤如下:
- 读取暴露因素GWAS数据
- 选取合适的工具变量(如有必要,需要进行clumping)
- 读取结局GWAS数据,提取上述的工具变量的SNP
- 对暴露因素与结局的GWAS数据进行预处理,使其格式统一
- 采用选中的MR方法进行分析
- 将结果可视化
- 散点图
- 森林图
- 漏斗图

1 读取暴露GWAS数据,选取工具变量
1.1 读取数据
对于暴露因素的GWAS数据,TwoSampleMR需要一个工具变量的data frame,每行对应一个SNP,至少需要4列,分别为:
SNP– rs IDbeta– The effect size. If the trait is binary then log(OR) should be usedse– The standard error of the effect sizeeffect_allele– The allele of the SNP which has the effect marked inbeta
其他可能有助于MR预处理或分析的列包括
other_allele– The non-effect alleleeaf– The effect allele frequencyPhenotype– The name of the phenotype for which the SNP has an effect
你也可以提供额外的信息(可选,对于分析非必须)
chr– Physical position of variant (chromosome)position– Physical position of variant (position)samplesize– Sample size for estimating the effect sizencase– Number of casesncontrol– Number of controlspval– The P-value for the SNP’s association with the exposureunits– The units in which the effects are presentedgene– The gene or other annotation for the the SNP
本教程将从本地文本文件中读取暴露GWAS数据:
本文使用的暴露因素的GWAS数据的前5行如下所示:
rsid,effect,SE,a1,a2,a1_freq,p-value,Units,Gene,n
rs10767664,0.19,0.030612245,A,T,0.78,5.00E-26,kg/m2,BDNF,225238
rs13078807,0.1,0.020408163,G,A,0.2,4.00E-11,kg/m2,CADM2,221431
rs1514175,0.07,0.020408163,A,G,0.43,8.00E-14,kg/m2,TNNI3K,207641
rs1558902,0.39,0.020408163,A,T,0.42,5.00E-120,kg/m2,FTO,222476
该示例数据随TwoSampleMR包一同下载,可以通过以下代码定位其位置,并读取:
## 定位数据位置,将路径赋予bmi2_file
bmi2_file <- system.file("extdata/bmi.csv", package="TwoSampleMR")
##读取数据
bmi_exp_dat <- read_exposure_data(
filename = bmi2_file,
sep = ",",
snp_col = "rsid",
beta_col = "effect",
se_col = "SE",
effect_allele_col = "a1",
other_allele_col = "a2",
eaf_col = "a1_freq",
pval_col = "p-value",
units_col = "Units",
gene_col = "Gene",
samplesize_col = "n"
)
1.2 Clumping
对于一个标准的两样本MR分析来说,我们需要确保工具变量之间是互相独立的(即不存在显著的LD),在读取完数据后我们应当对其进行LD Clumping,这里可以使用clump_data()函数,指定LD参考面板为EUR,r2的阈值为0.01,对数据进行Clumping。 该函数与OpenGWAS API进行交互,其存储了千人基因组中5个群体(EUR, SAS, EAS, AFR, AMR)的LD数据。对东亚人进行分析时指定pop = "EAS"即可。
bmi_exp_dat <- clump_data(bmi_exp_dat,clump_r2=0.01 ,pop = "EUR")
#> Please look at vignettes for options on running this locally if you need to run many instances of this command.
#> Clumping UVOe6Z, 30 variants, using EUR population reference
#> Removing 3 of 30 variants due to LD with other variants or absence from LD reference panel
2 读取结局GWAS数据,提取对应工具变量
结局数据可以像上一节暴露因素GWAS数据的读取一样从本地文件中读取,也可以从在线数据库中提取。因为上一节已经演示过本地数据的读取,在这里就演示从在线数据库中提取的方法, 使用extract_outcome_data函数,指定结局GWAS的id,与要提取的SNP的rsID,即可完成数据提取。
例如我们想分析BMI与冠心病(coronary heart disease)之间的因果关系,首先我们要查找冠心病GWAS在数据库中的id
2.1 查找相应的GWAS
使用available_outcomes()可以查看当前可用的所有表型的GWAS数据,然后使用grep1抓取关键字
ao <- available_outcomes()
#> API: public: <http://gwas-api.mrcieu.ac.uk/>
head(ao)
#> # A tibble: 6 x 22
#> id trait group_name year consortium author sex population unit
#> <chr> <chr> <chr> <int> <chr> <chr> <chr> <chr> <chr>
#> 1 ieu-b-~ Number o~ public 2021 MRC-IEU Clare~ Male~ European SD
#> 2 prot-a~ Leptin r~ public 2018 NA Sun BB Male~ European NA
#> 3 prot-a~ Adapter ~ public 2018 NA Sun BB Male~ European NA
#> 4 ukb-e-~ Impedanc~ public 2020 NA Pan-U~ Male~ Greater Midd~ NA
#> 5 prot-a~ Dual spe~ public 2018 NA Sun BB Male~ European NA
#> 6 eqtl-a~ ENSG0000~ public 2018 NA Vosa U Male~ European NA
#> # ... with 13 more variables: sample_size <int>, nsnp <int>, build <chr>,
#> # category <chr>, subcategory <chr>, ontology <chr>, note <chr>, mr <int>,
#> # pmid <int>, priority <int>, ncase <int>, ncontrol <int>, sd <dbl>
## 使用grep1抓取目标表型
ao[grepl("heart disease", ao$trait), ]
#> # A tibble: 28 x 22
#> id trait group_name year consortium author sex population unit
#> <chr> <chr> <chr> <int> <chr> <chr> <chr> <chr> <chr>
#> 1 ieu-a-8 Coronary h~ public 2011 CARDIoGRAM Schun~ Male~ European log ~
#> 2 finn-a~ Valvular h~ public 2020 NA NA Male~ European NA
#> 3 finn-a~ Other or i~ public 2020 NA NA Male~ European NA
#> 4 ukb-b-~ Diagnoses ~ public 2018 MRC-IEU Ben E~ Male~ European SD
#> 5 ukb-a-~ Diagnoses ~ public 2017 Neale Lab Neale Male~ European SD
#> 6 ukb-e-~ I25 Chroni~ public 2020 NA Pan-U~ Male~ South Asi~ NA
#> 7 ukb-b-~ Diagnoses ~ public 2018 MRC-IEU Ben E~ Male~ European SD
#> 8 finn-a~ Ischemic h~ public 2020 NA NA Male~ European NA
#> 9 finn-a~ Major coro~ public 2020 NA NA Male~ European NA
#> 10 finn-a~ Other hear~ public 2020 NA NA Male~ European NA
#> # ... with 18 more rows, and 13 more variables: sample_size <int>, nsnp <int>,
#> # build <chr>, category <chr>, subcategory <chr>, ontology <chr>, note <chr>,
#> # mr <int>, pmid <int>, priority <int>, ncase <int>, ncontrol <int>, sd <dbl>
查看完整结果后我们发现冠心病GWAS id为 ieu-a-7
当然可以直接进入数据库网站搜索 https://gwas.mrcieu.ac.uk/ ,简单直观
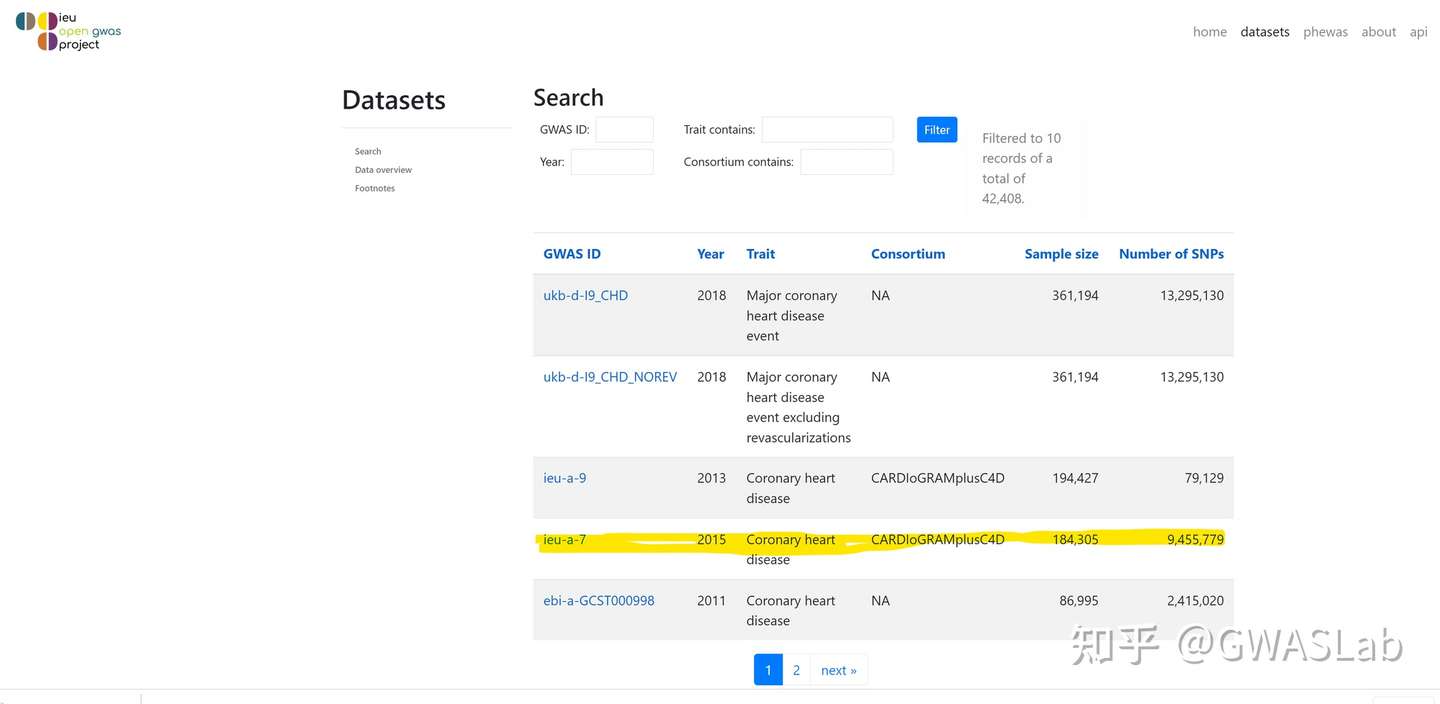
2.2 提取结局GWAS数据
指定GWAS id,指定要提取的SNP rsid (1中选取的工具变量),使用extract_outcome_data进行提取
chd_out_dat <- extract_outcome_data(
snps = bmi_exp_dat$SNP,
outcomes = 'ieu-a-7'
)
head(chd_out_dat )
A data.frame: 6 × 16
SNP chr pos beta.outcome se.outcome samplesize.outcome pval.outcome eaf.outcome effect_allele.outcome other_allele.outcome outcome id.outcome originalname.outcome outcome.deprecated mr_keep.outcome data_source.outcome
<chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <chr> <lgl> <chr>
1 rs887912 2 59302877 -0.021960 0.0111398 184305 0.04868780 0.731654 C T Coronary heart disease || id:ieu-a-7 ieu-a-7 Coronary heart disease Coronary heart disease || || TRUE igd
2 rs2112347 5 75015242 -0.005855 0.0096581 184305 0.54436400 0.401399 G T Coronary heart disease || id:ieu-a-7 ieu-a-7 Coronary heart disease Coronary heart disease || || TRUE igd
3 rs3817334 11 47650993 0.000355 0.0095386 184305 0.97031200 0.387831 T C Coronary heart disease || id:ieu-a-7 ieu-a-7 Coronary heart disease Coronary heart disease || || TRUE igd
4 rs1555543 1 96944797 0.002516 0.0093724 184305 0.78835500 0.558318 C A Coronary heart disease || id:ieu-a-7 ieu-a-7 Coronary heart disease Coronary heart disease || || TRUE igd
5 rs2815752 1 72812440 0.010402 0.0098384 184305 0.29038200 0.611282 A G Coronary heart disease || id:ieu-a-7 ieu-a-7 Coronary heart disease Coronary heart disease || || TRUE igd
6 rs10938397 4 45182527 0.030606 0.0093485 184305 0.00106079 0.416076 G A Coronary heart disease || id:ieu-a-7 ieu-a-7 Coronary heart disease Coronary heart disease || || TRUE igd
3 对数据进行预处理,使其效应等位与效应量保持统一
完成暴露因素与结局GWAS数据的提取后,我们要对其进行预处理,使其效应等位保持统一,可以使用harmonise_data函数,方便的完成处理:
dat <- harmonise_data(
exposure_dat = bmi_exp_dat,
outcome_dat = chd_out_dat
)
到这一步数据的准备工作就完成了,下一步可以开始分析
4 MR分析与可视化
4.1 MR分析
使用mr函数对处理好的数据dat 进行分析,结果如下
res <- mr(dat)
#> Analysing 'ieu-a-2' on 'ieu-a-7'
res
#> id.exposure id.outcome outcome
#> 1 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> 2 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> 3 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> 4 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> 5 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> exposure method nsnp b
#> 1 Body mass index || id:ieu-a-2 MR Egger 79 0.5024935
#> 2 Body mass index || id:ieu-a-2 Weighted median 79 0.3870065
#> 3 Body mass index || id:ieu-a-2 Inverse variance weighted 79 0.4459091
#> 4 Body mass index || id:ieu-a-2 Simple mode 79 0.3401554
#> 5 Body mass index || id:ieu-a-2 Weighted mode 79 0.3888249
#> se pval
#> 1 0.14396056 8.012590e-04
#> 2 0.07226598 8.541141e-08
#> 3 0.05898302 4.032020e-14
#> 4 0.15698810 3.330579e-02
#> 5 0.09240475 6.833558e-05
mr默认使用五种方法( MR Egger,Weighted median,Inverse variance weighted,Simple mode ,Weighted mode )
但我们可以额外指定方法,使用mr_method_list()查看当前支持的统计方法
mr_method_list()
obj name PubmedID Description use_by_default heterogeneity_test
<chr> <chr> <chr> <chr> <lgl> <lgl>
mr_wald_ratio Wald ratio TRUE FALSE
mr_two_sample_ml Maximum likelihood FALSE TRUE
mr_egger_regression MR Egger 26050253 TRUE TRUE
mr_egger_regression_bootstrap MR Egger (bootstrap) 26050253 FALSE FALSE
mr_simple_median Simple median FALSE FALSE
mr_weighted_median Weighted median TRUE FALSE
mr_penalised_weighted_median Penalised weighted median FALSE FALSE
mr_ivw Inverse variance weighted TRUE TRUE
mr_ivw_radial IVW radial FALSE TRUE
mr_ivw_mre Inverse variance weighted (multiplicative random effects) FALSE FALSE
mr_ivw_fe Inverse variance weighted (fixed effects) FALSE FALSE
mr_simple_mode Simple mode TRUE FALSE
mr_weighted_mode Weighted mode TRUE FALSE
mr_weighted_mode_nome Weighted mode (NOME) FALSE FALSE
mr_simple_mode_nome Simple mode (NOME) FALSE FALSE
mr_raps Robust adjusted profile score (RAPS) FALSE FALSE
mr_sign Sign concordance test Tests for concordance of signs between exposure and outcome FALSE FALSE
mr_uwr Unweighted regression Doesn't use any weights FALSE TRUE
在mr函数中,添加想要使用的方法即可:
mr(dat, method_list=c("mr_egger_regression", "mr_ivw"))
#> Analysing 'ieu-a-2' on 'ieu-a-7'
#> id.exposure id.outcome outcome
#> 1 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> 2 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> exposure method nsnp b
#> 1 Body mass index || id:ieu-a-2 MR Egger 79 0.5024935
#> 2 Body mass index || id:ieu-a-2 Inverse variance weighted 79 0.4459091
#> se pval
#> 1 0.14396056 8.01259e-04
#> 2 0.05898302 4.03202e-14
4.2 敏感性分析
4.2.1 异质性检验 mr_heterogeneity
mr_heterogeneity(dat)
#> id.exposure id.outcome outcome
#> 1 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> 2 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> exposure method Q Q_df
#> 1 Body mass index || id:ieu-a-2 MR Egger 143.3046 77
#> 2 Body mass index || id:ieu-a-2 Inverse variance weighted 143.6508 78
#> Q_pval
#> 1 6.841585e-06
#> 2 8.728420e-06
4.2.2 水平多效性检验 mr_pleiotropy_test
MR Egger 回归中的截距是反应水平多效性的一个有效指标,可以通过mr_pleiotropy_test计算
mr_pleiotropy_test(dat)
#> id.exposure id.outcome outcome
#> 1 ieu-a-2 ieu-a-7 Coronary heart disease || id:ieu-a-7
#> exposure egger_intercept se pval
#> 1 Body mass index || id:ieu-a-2 -0.001719304 0.003985962 0.6674266
4.3 可视化
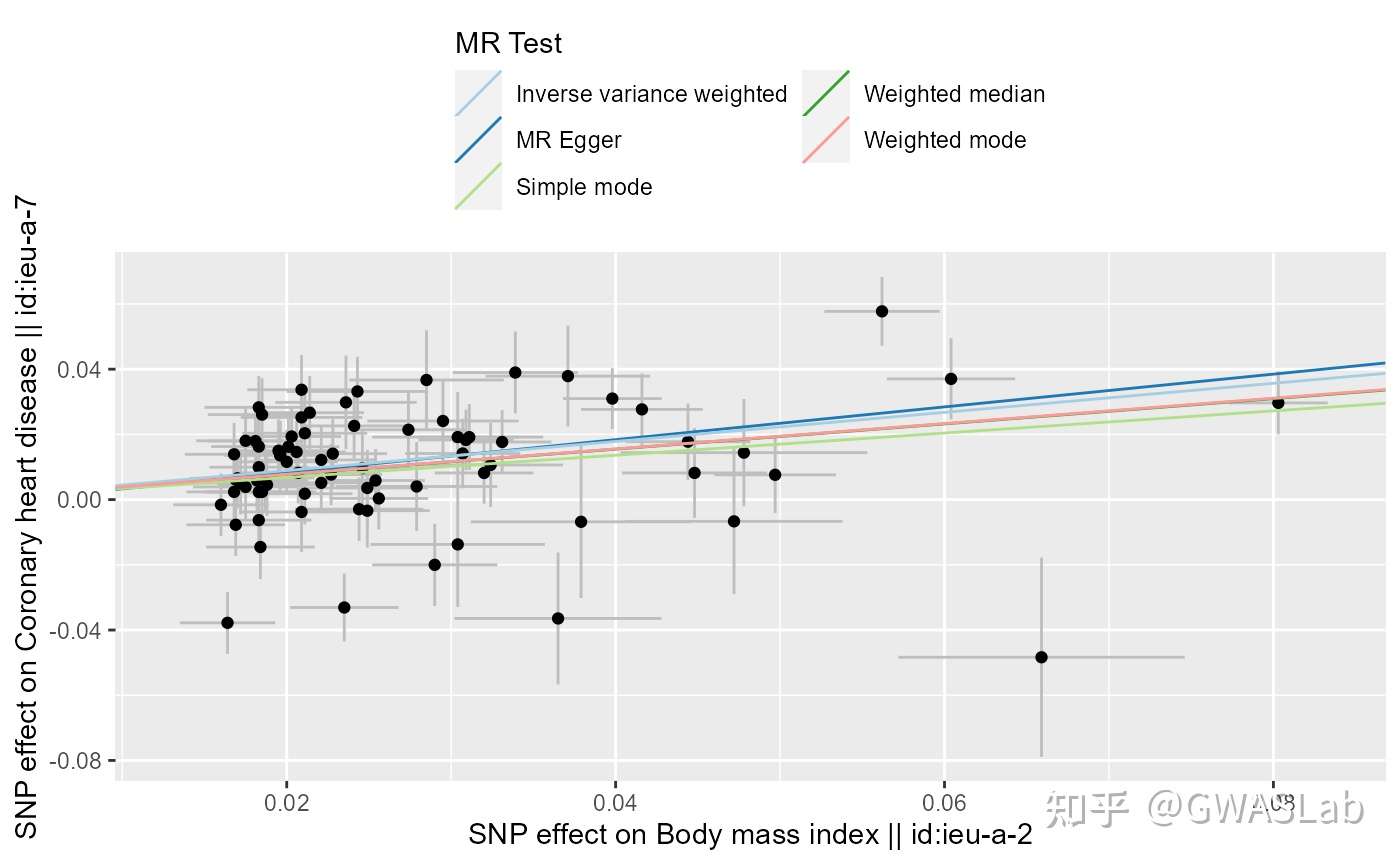
4.3.1 散点图
对上述的MR结果与输入数据,使用mr_scatter_plot函数绘制散点图
res <-mr(dat)
#> Analysing 'ieu-a-2' on 'ieu-a-7'
p1 <-mr_scatter_plot(res, dat)
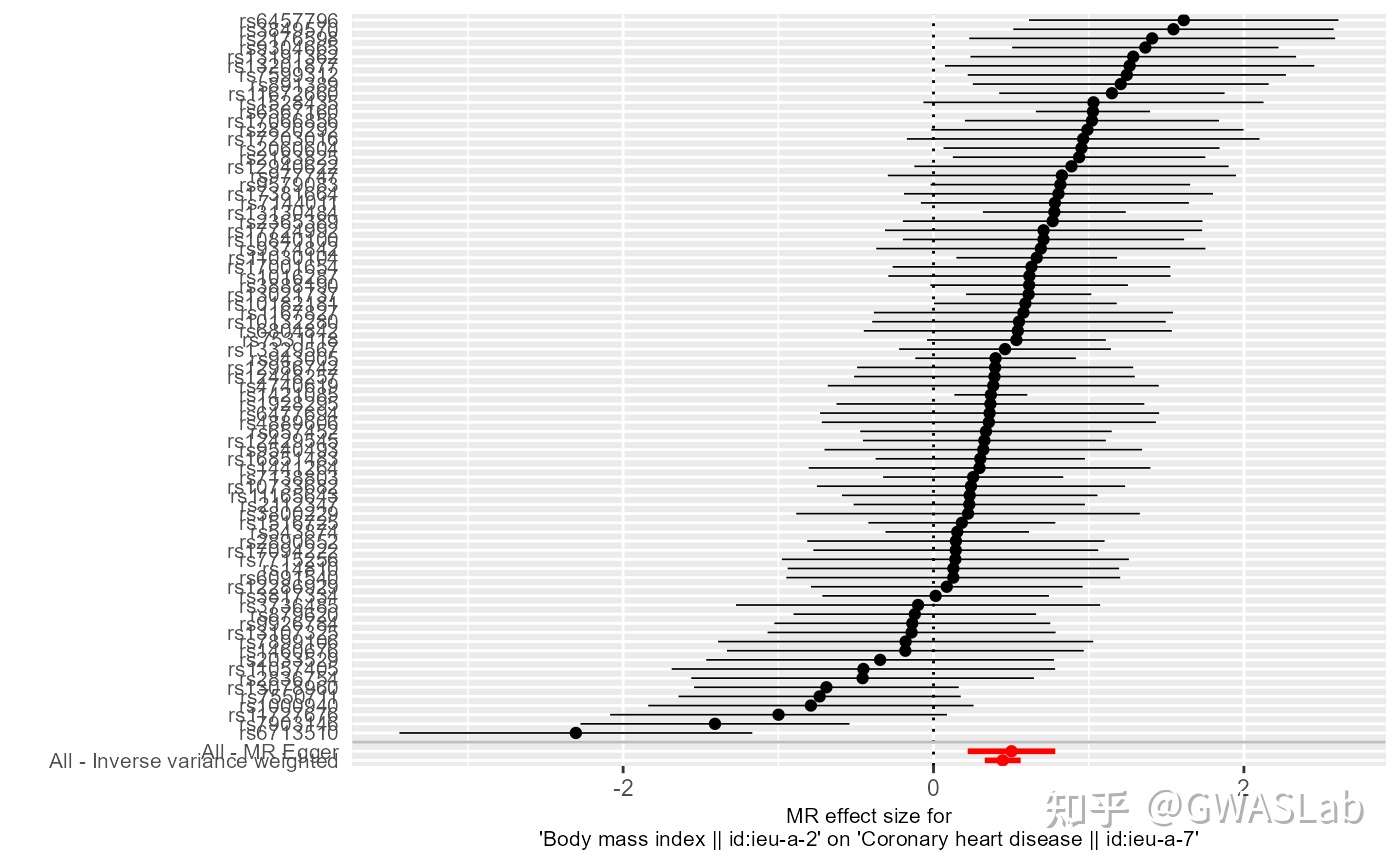
4.3.2 森林图
首先使用mr_singlesnp获取单个SNP的结果,然后使用mr_forest_plot绘制森林图
res_single <- mr_singlesnp(dat)
p2 <- mr_forest_plot(res_single)
p2[[1]]
#> Warning: Removed 1 rows containing missing values (geom_errorbarh).
#> Warning: Removed 1 rows containing missing values (geom_point).
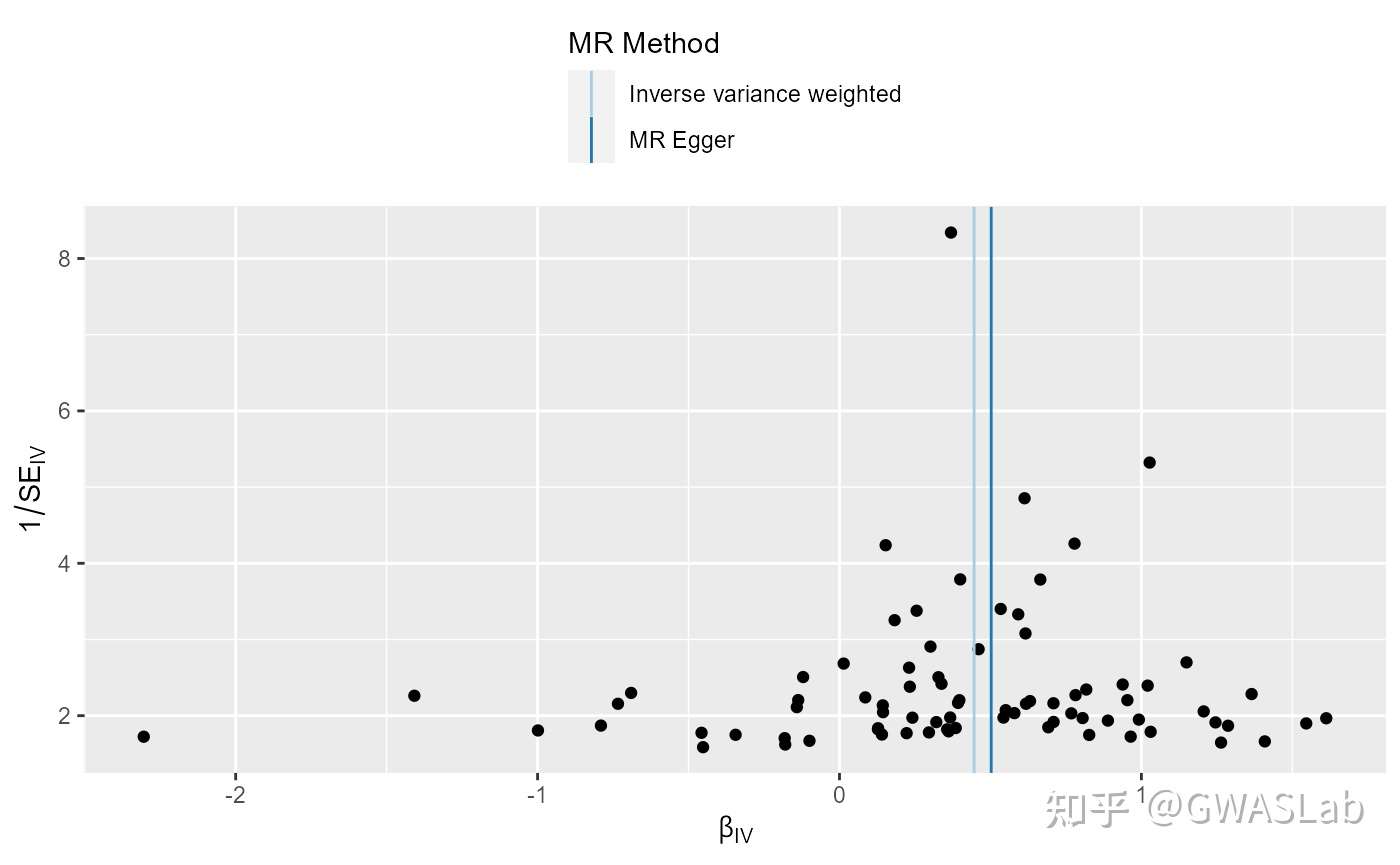
4.3.2 漏斗图
首先使用mr_singlesnp获取单个SNP的结果,然后使用mr_funnel_plot绘制漏斗图
res_single <- mr_singlesnp(dat)
p4 <- mr_funnel_plot(res_single)
p4[[1]]
参考:
Hemani G, Tilling K, Davey Smith G (2017) Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLOS Genetics 13(11): e1007081. https://doi.org/10.1371/journal.pgen.1007081
Hemani G, Zheng J, Elsworth B, Wade KH, Baird D, Haberland V, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Davey Smith G, Gaunt TR, Haycock PC, The MR-Base Collaboration. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018;7:e34408. doi: 10.7554/eLife.34408





