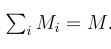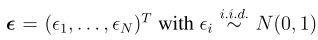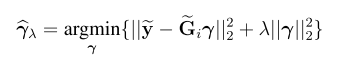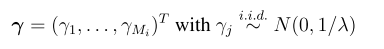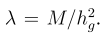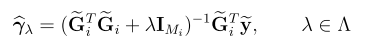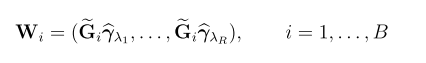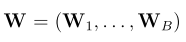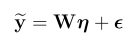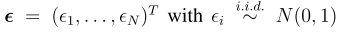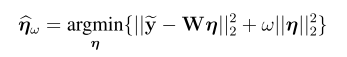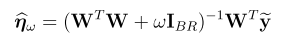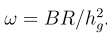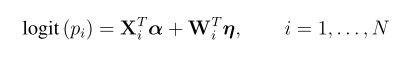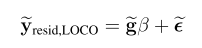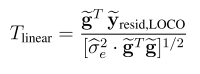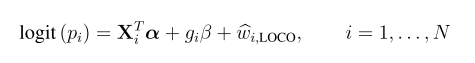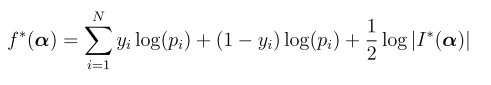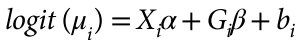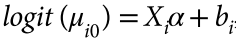关键字:conditional analysis,leading SNP, secondary casual variants , fine-mapping
GWAS研究中,对于某个复杂表型,我们会发现很多显著关联的基因座(loci),每个基因座里有若干显著的SNP,这些SNP通常处于LD。这时我们就会面临一个问题,这个基因座里显著的SNP单单因为与leading SNP(该loci里P值最低的SNP)连锁不平衡而显著,还是因为这个SNP本身就与就与表型相关联。这时就应该进行条件分析(conditional analysis),实际上是一种fine-mapping(对casual SNP 致病SNP的精确定位)的方法,目的是确认是否存在次要的因果SNP secondary causal variants。
一般来说进行条件分析的方法很简单,
1.从GWAS结果中抽出每个关联基因座的leading SNP,
2.将该leading SNP作为协变量加入检验模型
3.再次进行关联检验,确认 关联基因座里除 leading SNP 以外还是否有次要的致病SNP secondary causal variants。
例如下图所示的情况:

图1:针对橙色圈中的leading SNP (该loci里p值最低),进行条件分析后的曼哈顿图。A:该loci只有一个信号,表示没有 secondary causal variants ,之所以有多个显著snp是因为与leading SNP处于连锁不平衡。B:除了leading SNP 还有其他信号,表示这个loci还存在 secondary causal variants 。
通常conditional analysis的功能已经集成在关联检验的软件中(如PLINK,SAIGE),我们只需要提供lead snp的 id 或是 位置,就可以进行条件分析了。
例如:
PLINK中的 –condition 选项,也可以使用 –condition–list(同时将多个SNP作为covariate纳入检验模型)
SAIGE 第二步中,也提供了 –condition 选项 ,可以对单个或多个snp进行条件分析
参考:
Strategies for fine-mapping complex traits.
https://www.cog-genomics.org/plink/1.9/assoc
https://github.com/weizhouUMICH/SAIGE/wiki/Genetic-association-tests-using-SAIGE