本文内容:
- 下载PGS分数文件
- 第一步 计算每条染色体的原始分数
- 第二步 将各个染色体的结果提取
- 第三步 将分数进行简单加和
本文主要演示分染色体计算PRS并求和的过程。
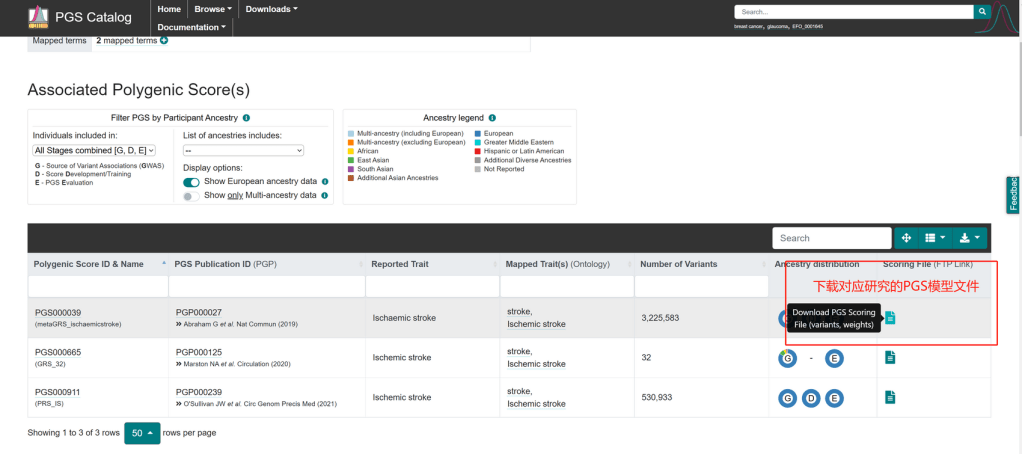
下载PGS分数文件 Download PGS score
演示PGS分数文件下载自PGScatalog

下载harmonise后的文件:
wget https://ftp.ebi.ac.uk/pub/databases/spot/pgs/scores/PGS000012/ScoringFiles/Harmonized/PGS000012_hmPOS_GRCh37.txt.gz
gunzip PGS000012_hmPOS_GRCh37.txt.gz
head PGS000012_hmPOS_GRCh37.txt
###PGS CATALOG SCORING FILE - see <https://www.pgscatalog.org/downloads/#dl_ftp_scoring> for additional information
#format_version=2.0
##POLYGENIC SCORE (PGS) INFORMATION
#pgs_id=PGS000012
#pgs_name=GRS49K
#trait_reported=Coronary artery disease
#trait_mapped=coronary artery disease
#trait_efo=EFO_0001645
#genome_build=hg19
#variants_number=49310
#下载后去除文件头部的comment,方便Plink2使用
awk '$1!~/#/{print $0}' PGS000012_hmPOS_GRCh37.txt > PGS000012_hmPOS_GRCh37_plink2.txt
head PGS000012_hmPOS_GRCh37_plink2.txt
rsID effect_allele effect_weight hm_source hm_rsID hm_chr hm_pos hm_inferOtherAllele
rs1333045 C 0.187251 ENSEMBL rs1333045 9 22119195 T
rs1537370 T 0.17296 ENSEMBL rs1537370 9 22084310 C
rs9863247 T 0.136328 ENSEMBL rs9863247 3 161125373 C
rs11203077 T 0.131725 ENSEMBL rs11203077 10 91097085 G
rs10268558 C 0.12966 ENSEMBL rs10268558 7 18924927 T
rs7148203 C 0.128366 ENSEMBL rs7148203 14 45062275 T
rs12747328 T 0.127016 ENSEMBL rs12747328 1 159429702 C
rs17810947 G 0.126626 ENSEMBL rs17810947 18 70754886 A/T
rs2412710 A 0.125677 ENSEMBL rs2412710 15 42683787 G
基因组文件使用 https://choishingwan.github.io/PRS-Tutorial/plink/ 所提供的的EUR文件
EUR.QC.bed
EUR.QC.bim
EUR.QC.fam
注:本教程计算结果无实际意义,仅为演示计算过程使用。
第一步 计算每条染色体的原始分数 step1 calculate the raw scores for each chromosome
score=PGS000012_hmPOS_GRCh37_plink2.txt
for chr in $(seq 1 22)
do
plink2 \
--bfile ./EUR.QC \
--score ${score} 1 2 3 header list-variants cols=+scoresums \
--chr ${chr} \
--out EUR_CAD.chr${chr}
done
—score 后跟 分数文件路径, 并指定 文件的 ID, effect allele 以及 effect_weight的列数。
header :分数文件有header,计算时会自动跳过第一行
list-variants : 将计算所用的所有variant ID写进plink2.sscore.vars
—chr 指定所计算的染色体编号
注1: 这里要加上cols=+scoresums 来获取原始结果,默认输出的仅为平均后的结果
--score applies one or more linear scoring systems to each sample, and reports results to plink2.sscore. More precisely, if G is the full genotype/dosage matrix (rows = alleles, columns = samples) and a is a scoring-system vector with one coefficient per allele, --score computes the vector-matrix product aTG, and then divides by the number of variants when reporting score-averages.
注2:有时候分数文件中不包括某些染色体的variants,所以有些染色会计算失败,求和时应注意
所得文件
EUR_CAD.chr10.log EUR_CAD.chr15.log EUR_CAD.chr1.log EUR_CAD.chr3.log EUR_CAD.chr8.log
EUR_CAD.chr10.sscore EUR_CAD.chr15.sscore EUR_CAD.chr1.sscore EUR_CAD.chr3.sscore EUR_CAD.chr8.sscore
EUR_CAD.chr10.sscore.vars EUR_CAD.chr15.sscore.vars EUR_CAD.chr1.sscore.vars EUR_CAD.chr3.sscore.vars EUR_CAD.chr8.sscore.vars
EUR_CAD.chr11.log EUR_CAD.chr16.log EUR_CAD.chr20.log EUR_CAD.chr4.log EUR_CAD.chr9.log
EUR_CAD.chr11.sscore EUR_CAD.chr16.sscore EUR_CAD.chr20.sscore EUR_CAD.chr4.sscore EUR_CAD.chr9.sscore
EUR_CAD.chr11.sscore.vars EUR_CAD.chr16.sscore.vars EUR_CAD.chr20.sscore.vars EUR_CAD.chr4.sscore.vars EUR_CAD.chr9.sscore.vars
EUR_CAD.chr12.log EUR_CAD.chr17.log EUR_CAD.chr21.log EUR_CAD.chr5.log
EUR_CAD.chr12.sscore EUR_CAD.chr17.sscore EUR_CAD.chr21.sscore EUR_CAD.chr5.sscore
EUR_CAD.chr12.sscore.vars EUR_CAD.chr17.sscore.vars EUR_CAD.chr21.sscore.vars EUR_CAD.chr5.sscore.vars
EUR_CAD.chr13.log EUR_CAD.chr18.log EUR_CAD.chr22.log EUR_CAD.chr6.log
EUR_CAD.chr13.sscore EUR_CAD.chr18.sscore EUR_CAD.chr22.sscore EUR_CAD.chr6.sscore
EUR_CAD.chr13.sscore.vars EUR_CAD.chr18.sscore.vars EUR_CAD.chr22.sscore.vars EUR_CAD.chr6.sscore.vars
EUR_CAD.chr14.log EUR_CAD.chr19.log EUR_CAD.chr2.log EUR_CAD.chr7.log
EUR_CAD.chr14.sscore EUR_CAD.chr19.sscore EUR_CAD.chr2.sscore EUR_CAD.chr7.sscore
EUR_CAD.chr14.sscore.vars EUR_CAD.chr19.sscore.vars EUR_CAD.chr2.sscore.vars EUR_CAD.chr7.sscore.vars
分数文件内容
head EUR_CAD.chr1.sscore
#FID IID ALLELE_CT NAMED_ALLELE_DOSAGE_SUM SCORE1_AVG SCORE1_SUM
HG00096 HG00096 1046 486 0.00757595 7.92444
HG00097 HG00097 1046 484 0.00783352 8.19386
HG00099 HG00099 1046 505 0.00798495 8.35226
HG00101 HG00101 1046 491 0.00791584 8.27997
HG00102 HG00102 1046 474 0.00769178 8.04561
HG00103 HG00103 1046 502 0.00799328 8.36097
HG00105 HG00105 1046 500 0.00793847 8.30364
HG00107 HG00107 1046 468 0.00770316 8.0575
HG00108 HG00108 1046 469 0.00732639 7.66341
分染色体的计算的目的是,实际操作中,通常会使用巨大的pgen或bgen进行计算,合并文件并不现实, 这是一般需要分染色体计算并手动求和:
for chr in $(seq 1 22)
do
plink2 \
--pfile chr${chr} \
--score ${score} 1 2 3 header list-variants cols=+scoresums \
--out chr${chr}
done
第二步 将各个染色体的结果提取 step2 extract raw scores into a single file
scoreFile=EUR_CAD
count=1
for chr in $(seq 1 22)
do
file=${scoreFile}.chr${chr}.sscore
if [ -s "$file" ] ; then
if [ ${count} -eq 1 ] ;then
echo "ID chr${chr}" > ./${scoreFile}.chrALL.score
awk 'NR>1{print $1,$6}' ./${file} >> ./${scoreFile}.chrALL.score
else
paste ./${scoreFile}.chrALL.score <(cut -f6 ./${file} | awk -v chr=chr${chr} 'NR==1 {$0=chr} 1') >./${scoreFile}.temp&&mv ./${scoreFile}.temp ./${scoreFile}.chrALL.score
fi
count=`expr $count + 1`
fi
done
所得文件
head EUR_CAD.chrALL.score
ID chr1 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chr20 chr21 chr22
HG00096 7.92444 6.41628 4.9392 3.9937 5.08024 7.50241 4.61922 3.59464 4.24131 4.56312 5.03334 5.55227 2.95436 2.32917 3.39488 3.61625 3.40913 2.02032 1.95575 2.74394 0.79493 1.33408
HG00097 8.19386 6.33598 5.0713 4.0514 4.92887 7.30242 4.19193 3.66247 4.514 4.42716 5.26378 5.11748 2.7589 2.47905 3.82365 3.83448 3.44519 2.09788 1.80506 2.7079 0.811121 1.27481
HG00099 8.35226 6.61542 5.199 3.94708 4.88824 7.72775 4.31098 3.58159 4.30785 4.81232 5.2327 6.05063 2.8803 2.3651 3.36827 3.54378 3.29725 2.08449 1.88188 2.52587 0.767397 1.21001
HG00101 8.27997 6.13397 5.36465 4.04592 4.88809 7.86323 4.49118 3.60872 4.27056 4.6258 5.42121 5.26046 2.70375 2.52093 3.56581 3.45794 3.30534 2.1327 1.70916 2.66579 0.818805 1.3808
HG00102 8.04561 6.32975 5.20324 3.86051 4.67724 7.78206 4.69662 3.38095 4.40059 4.67174 5.2312 5.04913 2.69084 2.48935 3.62454 3.61557 3.3044 2.12043 1.96028 2.82496 0.808453 1.46265
HG00103 8.36097 6.1202 5.31402 4.24136 4.91736 7.34715 4.55809 3.60716 4.15705 4.66235 5.04477 5.53387 2.70537 2.36537 3.76842 3.64108 3.33273 2.11081 2.00393 2.68089 0.763437 1.32735
HG00105 8.30364 6.13937 5.15716 4.19656 5.06392 7.8889 4.33995 3.78158 4.24975 4.58436 5.32105 5.72797 2.84577 2.43968 3.39822 3.60722 3.44641 1.9986 1.775 2.7736 0.793725 1.41736
HG00107 8.0575 6.51259 5.19973 4.06067 4.78711 7.14954 4.66686 3.81154 4.42637 4.45144 5.26257 5.46242 2.82289 2.28362 3.33811 3.58244 3.25041 2.11758 2.00922 2.70618 0.917279 1.43502
HG00108 7.66341 6.18669 4.98761 4.05088 4.7294 7.39041 4.08688 3.53173 4.43728 4.80822 5.02485 5.52436 3.12156 2.35199 3.54311 3.49076 2.97043 2.12883 1.91326 2.51056 0.817221 1.47096
第三步 将分数进行简单加和 step3 combine the scores using awk
scoreFile=EUR_CAD
#sum the scores from each chr
awk 'BEGIN{t=0}
{if(NR==1){
print "ID","score"
}
else{
for(i=2;i<=NF;i++) t+=$i
print $1,t
}
}' ./${scoreFile}.chrALL.score > ./${scoreFile}.summary.score
所得文件
head EUR_CAD.summary.score
ID score
HG00096 88.013
HG00097 176.112
HG00099 265.062
HG00101 353.577
HG00102 441.807
HG00103 530.37
HG00105 619.62
HG00107 707.931
HG00108 794.672
求和完成,获得了原始分数(没有进行任何操作,没有标准化,没有平均,只是所有variant的效应量与dosage乘积的和),可以用于后续PRS分析
参考:
https://www.cog-genomics.org/plink/2.0/score