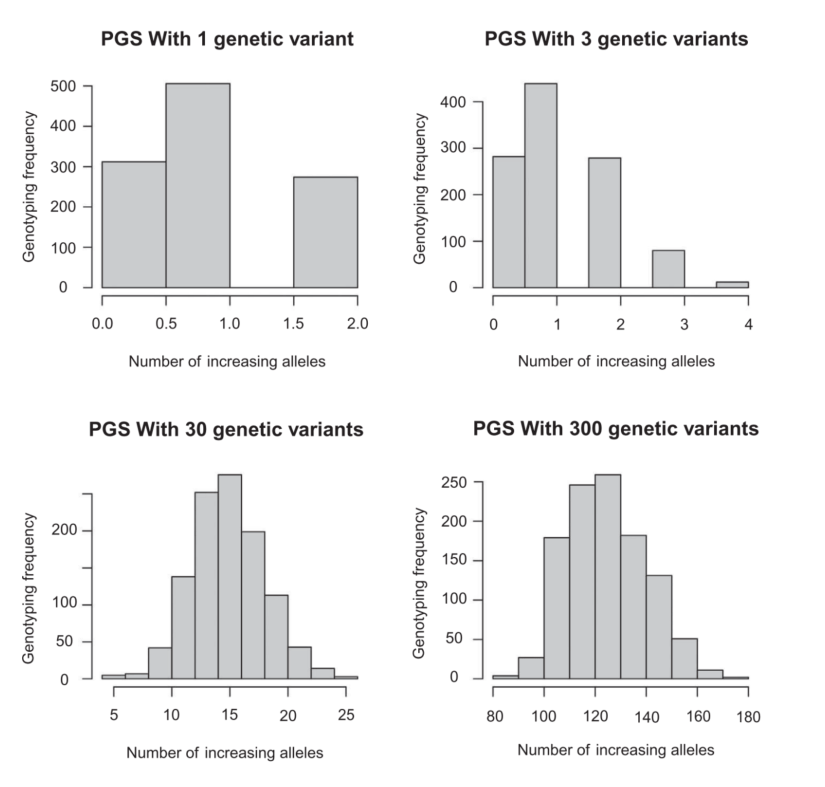
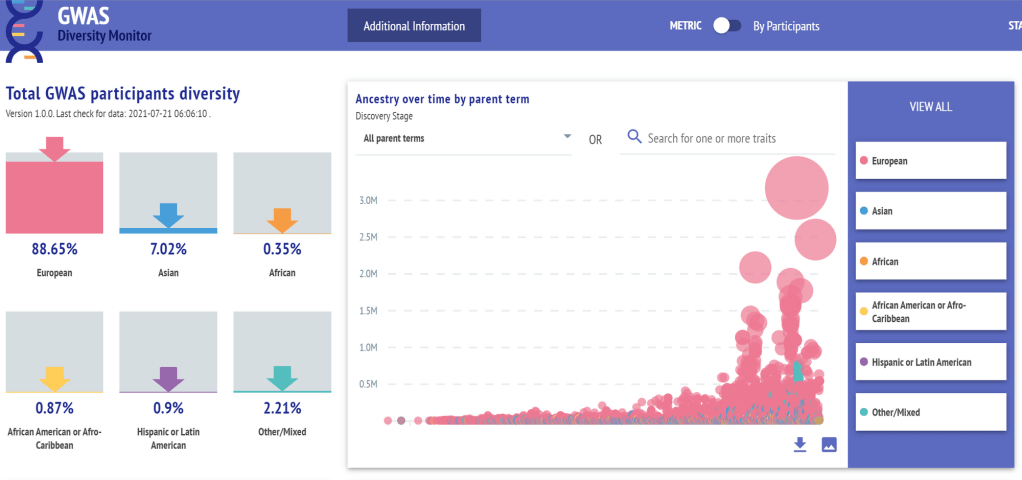
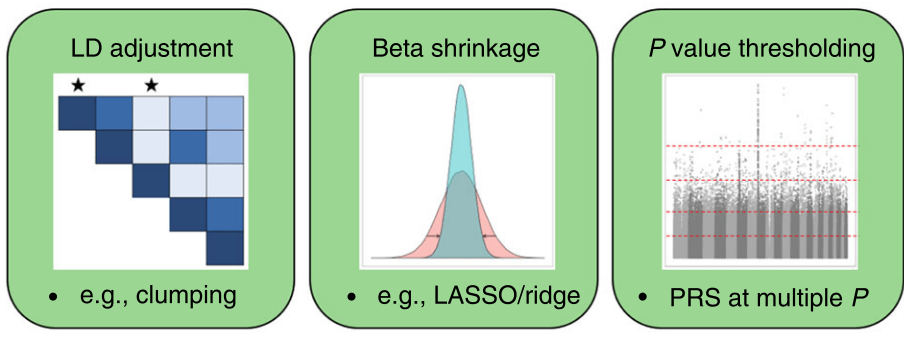
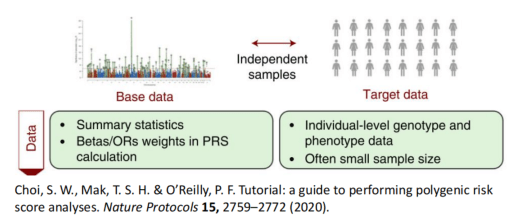
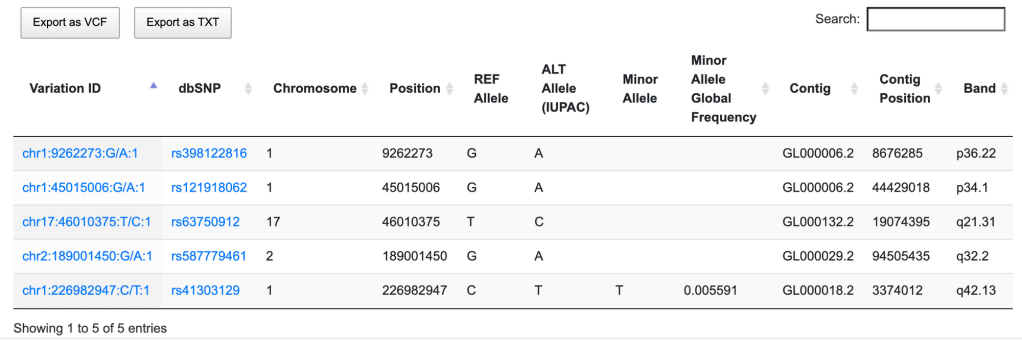
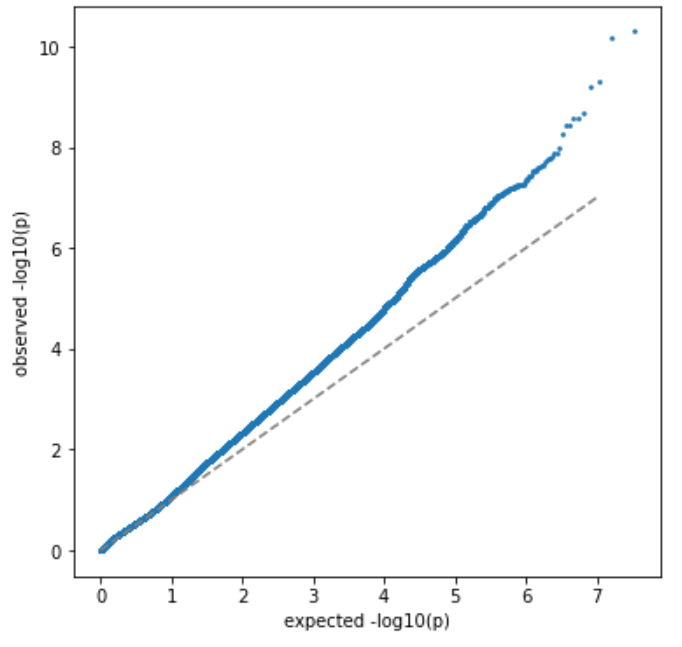
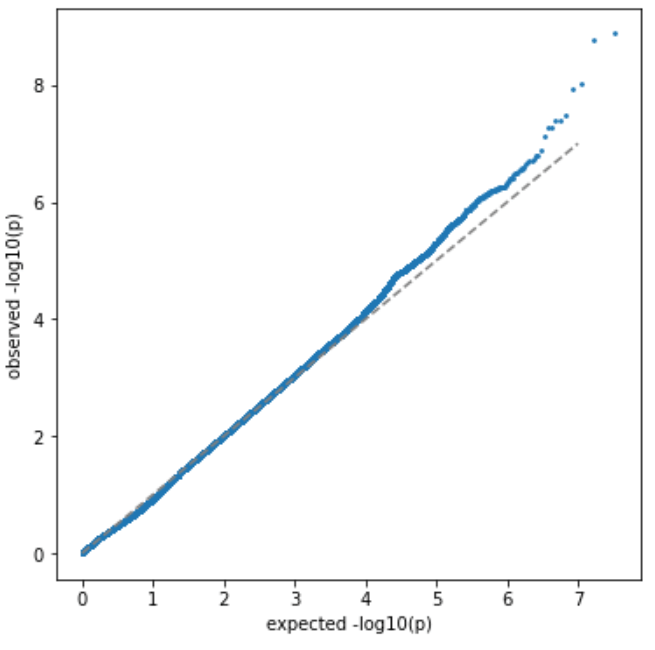
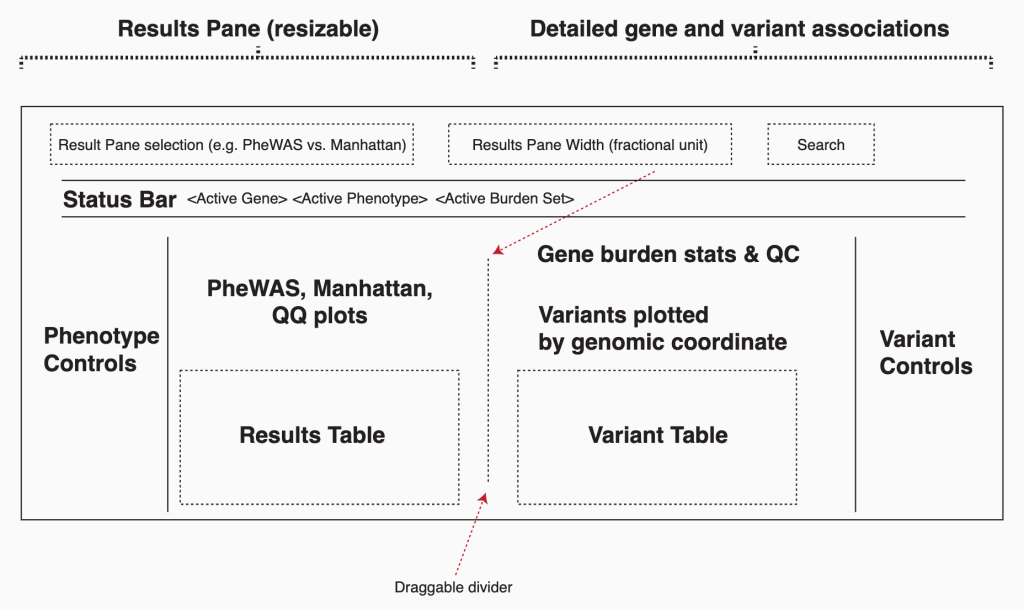
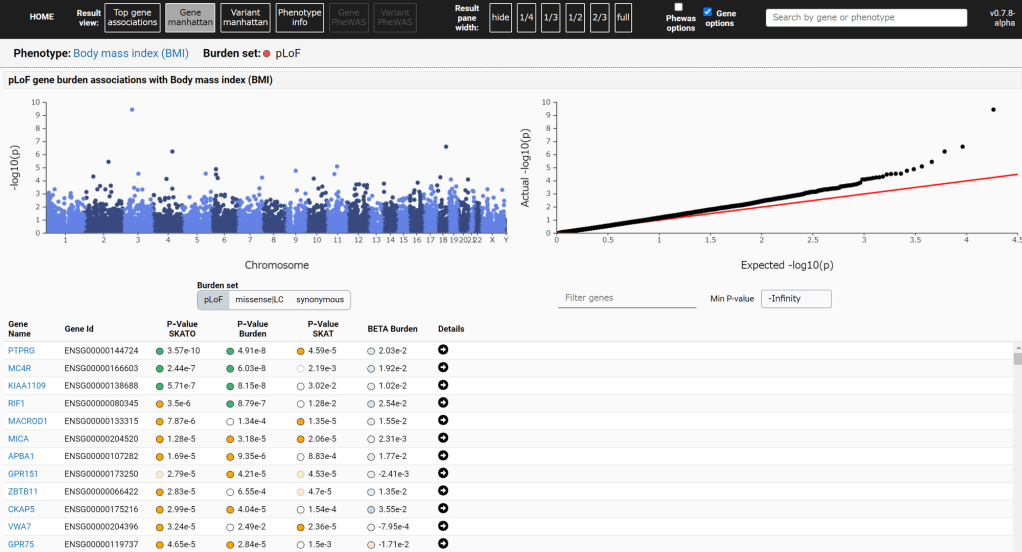
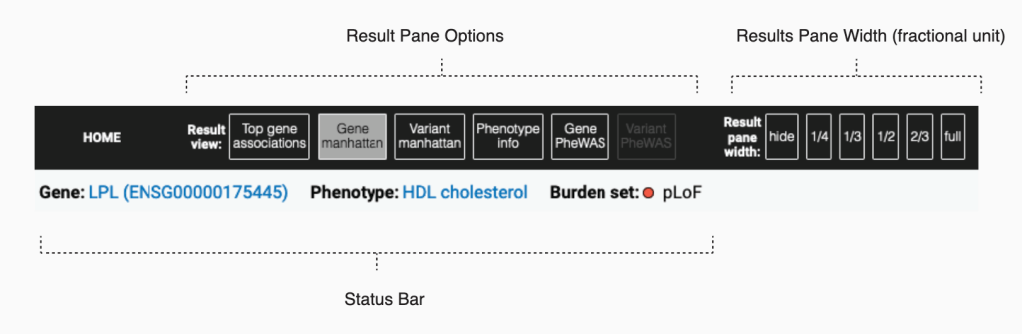
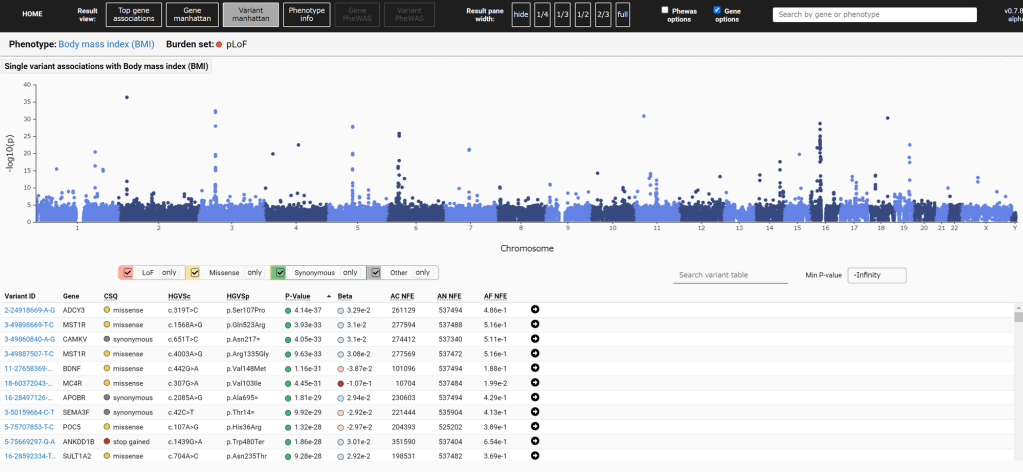
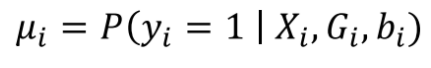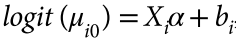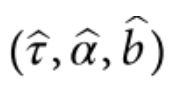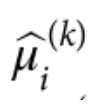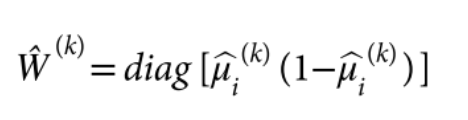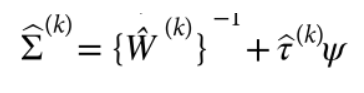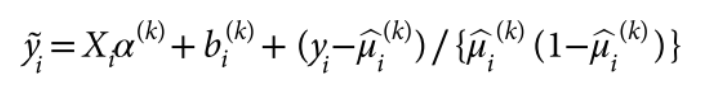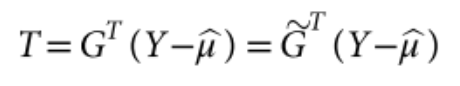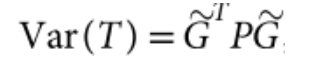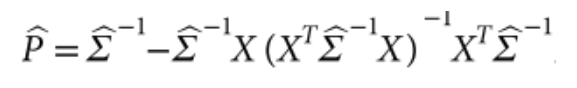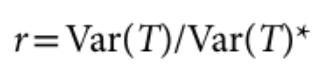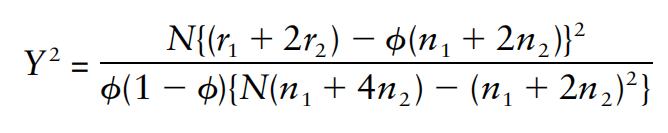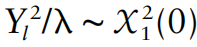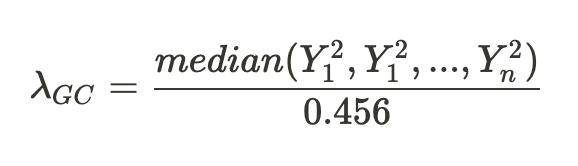第一组 频率上的 major 与 minor allele
第二组 参考基因组的 reference (ref) 与 alternative (alt) allele
第三组 关联检验的 reference (non-risk 或者 non-effect)与 risk/effect allele
首先第一组概念 major 与 minor allele
major allele 与 minor allele 通常针对某一大小确定的特定群体而言,频率最高的allele为该群体的major allele, 频率次高的为 minor allele,对于最常见的bi-allelic SNP来说,两个allele频率一高一低,就是这个群体中这个snp的major和minor allele,对于tri- 或者quad-allelic SNP (位点有三种或四种碱基的SNP)而言,minor allele则是频率第二高的那个allele
与plink1.9不同,plink2使用的概念则是reference 与 alternative allele,进行操作时不会自动依据频率而改变ref与alt的排序,使用plink2 的—frq选项计算频率,你会发现输出的文件中是alternative allele frequency (不是MAF),取值范围为[0,1]。
head Height.QC
CHR BP SNP A1 A2 N SE P OR INFO MAF
1 756604 rs3131962 A G 388028 0.00301666 0.483171 0.997886915712650.890557941364774 0.369389592764921
1 768448 rs12562034 A G 388028 0.00329472 0.834808 1.000687316093530.895893511351165 0.336845754096289
1 779322 rs4040617 G A 388028 0.00303344 0.42897 0.997603556067569 0.897508290615237 0.377368010940814
1 801536 rs79373928 G T 388028 0.00841324 0.808999 1.002035699227930.908962856432993 0.483212245374095
1 808631 rs11240779 G A 388028 0.00242821 0.590265 1.001308325111540.893212523690488 0.450409558999587
1 809876 rs57181708 G A 388028 0.00336785 0.71475 1.00123165786833 0.923557624081969 0.499743932656759
1 835499 rs4422948 G A 388028 0.0023758 0.710884 0.999119752645200.906437735120596 0.481016005816168
1 838555 rs4970383 A C 388028 0.00235773 0.150993 0.996619945289750.907716506801574 0.327164029672754
1 840753 rs4970382 C T 388028 0.00207377 0.199967 0.997345678956140.914602590137255 0.498936220426316
可以利用简单的R脚本进行转换,在这里我们在文件的最后一列后添加转换后的beta
dat <- read.table(gzfile("Height.QC.gz"), header=T)
dat$BETA <- log(dat$OR)
write.table(dat, "Height.QC.Transformed", quote=F, row.names=F)
q() # exit R
将OR转换成beta后
head Height.QC.Transformed
CHR BP SNP A1 A2 N SE P OR INFO MAF BETA
1 756604 rs3131962 A G 388028 0.00301666 0.483171 0.997886915712657 0.890557941364774 0.369389592764921 -0.00211532000000048
1 768448 rs12562034 A G 388028 0.00329472 0.834808 1.00068731609353 0.895893511351165 0.336845754096289 0.000687079999998082
1 779322 rs4040617 G A 388028 0.00303344 0.42897 0.997603556067569 0.897508290615237 0.377368010940814 -0.00239932000000021
1 801536 rs79373928 G T 388028 0.00841324 0.808999 1.00203569922793 0.908962856432993 0.483212245374095 0.00203362999999797
1 808631 rs11240779 G A 388028 0.00242821 0.590265 1.00130832511154 0.893212523690488 0.450409558999587 0.00130747000000259
1 809876 rs57181708 G A 388028 0.00336785 0.71475 1.00123165786833 0.923557624081969 0.499743932656759 0.00123090000000354
1 835499 rs4422948 G A 388028 0.0023758 0.710884 0.999119752645202 0.906437735120596 0.481016005816168 -0.000880634999999921
1 838555 rs4970383 A C 388028 0.00235773 0.150993 0.996619945289758 0.907716506801574 0.327164029672754 -0.0033857799999997
1 840753 rs4970382 C T 388028 0.00207377 0.199967 0.99734567895614 0.914602590137255 0.498936220426316 -0.00265785000000019
head EUR.clumped
CHR F SNP BP P TOTAL NSIG S05 S01 S001 S0001 SP2
6 1 rs3134762 31210866 4.52e-165 180 20 0 0 0 160 rs2523898(1),rs13210132(1),rs28732080(1),rs28732081(1),rs28732082(1),rs2517552(1),rs2517546(1),rs2844647(1),rs2517537(1),rs2844645(1),rs2523857(1),rs2517527(1),rs3131788(1),rs3132564(1),rs3130955(1),rs3130544(1),rs2535296(1),rs2517448(1),rs6457327(1),rs28732096(1),rs2233956(1),rs1265048(1),rs3130985(1),rs13201757(1),rs6909321(1),rs3130557(1),rs17196961(1),rs9501055(1),rs13200022(1),rs3130573(1),rs28732101(1),rs1265095(1),rs2233940(1),rs130079(1),rs746647(1),rs2240064(1),rs3131012(1)...
p.threshold <- c(0.001,0.05,0.1,0.2,0.3,0.4,0.5)
# Read in the phenotype file
phenotype <- read.table("EUR.height", header=T)
# Read in the PCs
pcs <- read.table("EUR.eigenvec", header=F)
# The default output from plink does not include a header
# To make things simple, we will add the appropriate headers
# (1:6 because there are 6 PCs)
colnames(pcs) <- c("FID", "IID", paste0("PC",1:6))
# Read in the covariates (here, it is sex)
covariate <- read.table("EUR.cov", header=T)
# Now merge the files
pheno <- merge(merge(phenotype, covariate, by=c("FID", "IID")), pcs, by=c("FID","IID"))
# We can then calculate the null model (model with PRS) using a linear regression
# (as height is quantitative)
null.model <- lm(Height~., data=pheno[,!colnames(pheno)%in%c("FID","IID")])
# And the R2 of the null model is
null.r2 <- summary(null.model)$r.squared
prs.result <- NULL
for(i in p.threshold){
# Go through each p-value threshold
prs <- read.table(paste0("EUR.",i,".profile"), header=T)
# Merge the prs with the phenotype matrix
# We only want the FID, IID and PRS from the PRS file, therefore we only select the
# relevant columns
pheno.prs <- merge(pheno, prs[,c("FID","IID", "SCORE")], by=c("FID", "IID"))
# Now perform a linear regression on Height with PRS and the covariates
# ignoring the FID and IID from our model
model <- lm(Height~., data=pheno.prs[,!colnames(pheno.prs)%in%c("FID","IID")])
# model R2 is obtained as
model.r2 <- summary(model)$r.squared
# R2 of PRS is simply calculated as the model R2 minus the null R2
prs.r2 <- model.r2-null.r2
# We can also obtain the coeffcient and p-value of association of PRS as follow
prs.coef <- summary(model)$coeff["SCORE",]
prs.beta <- as.numeric(prs.coef[1])
prs.se <- as.numeric(prs.coef[2])
prs.p <- as.numeric(prs.coef[4])
# We can then store the results
prs.result <- rbind(prs.result, data.frame(Threshold=i, R2=prs.r2, P=prs.p, BETA=prs.beta,SE=prs.se))
}
# Best result is:
prs.result[which.max(prs.result$R2),]
q() # exit R
执行后就可以看到本次PRS计算中,最优的阈值为0.3
Rscript bestfit.R
Threshold R2 P BETA SE
5 0.3 0.1638468 1.025827e-25 47343.32 4248.152
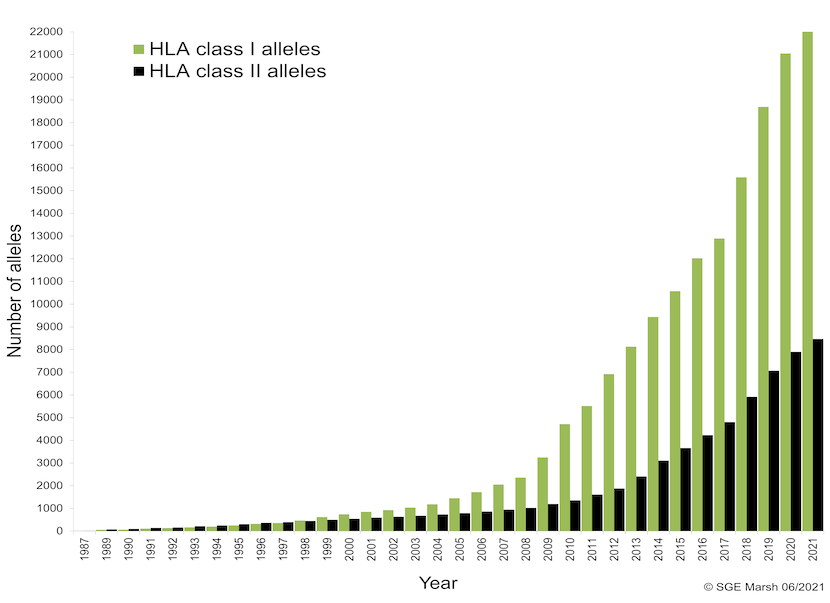
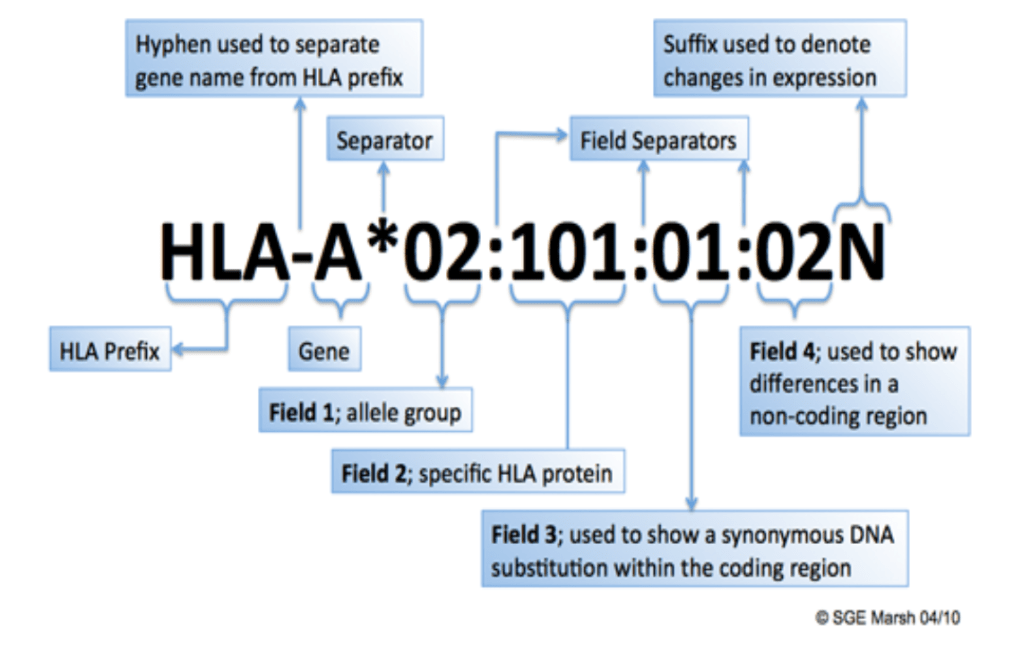
WHO的 Nomenclature Committee for Factors of the HLA System 为了区分编码HLA分子的HLA等位基因,于1987年开始施行4位数字的命名方法。后在2002年,由于4位数字的命名法已经不再适应该领域的快速发展,遂将HLA-A*02 群改为 A*92,将HLA-B15改为B95进行管理。但近年随着等位基因数的不断增加,其他的抗原群也难以在现行命名规则下管理。于是在2010年,发布了现行的HLA等位基因命名规则。这次变更后,使用冒号“:”分隔表示不同HLA等位基因的4个数字区域,并取消了固定位数的限制。
・第 3 区域:不伴随氨基酸变异的等位基因(同义置换:synonymous DNA substitution)(例:A*02:01:02,A*02:01:03,A*02:01:04 等) ・第 4 区域:非编码区域伴随碱基置换的等位基因(例:A*02:01:01:01,A*02:01:01:02L,A*02:01:01:03 等)
一些特殊标记:
存在两种以上等位基因无法判定时使用 / 分隔,例如 :HLA-DRB1*15/16
无法判定的等位基因过多时,使用+表示省略,例如:HLA-DRB1*15:01/16:01/+
表示肽链结合区(HLA class I:exon2 和3,HLA class II :exon2)的不确定性时,氨基酸序列(P),或是碱基序列(G)一致的等位基因,在末尾添加P或G表示。例如:A02:01:01:01/02:01:01:02L/02:01:01:03/02:01:02/02:01:03/02:01:04/02:01:05/02:01:06/02:01:07/02:01:08/02:01:09/02:01:10/02:01:11/02:01:12/02:01:13/02:01:14/02:01:15/02:01:17/02:01:18/02:01:19/02:01:21/02:01:22/02:09/02:66/02:75/02:89/02:97/02:132/02:134/02:140 可以表示为 A02:01P
Choi, S. W., Mak, T. S. H. & O’Reilly, P. F. Tutorial: a guide to performing polygenic risk score analyses. Nature Protocols 15, 2759–2772 (2020).
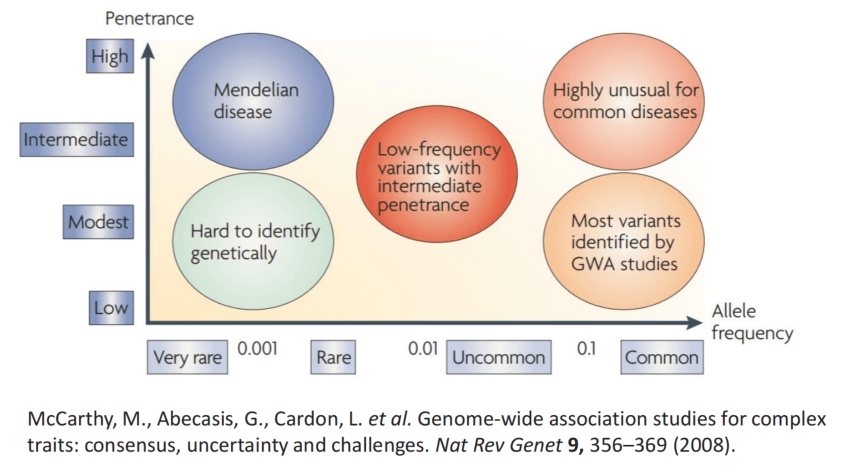
McCarthy, M., Abecasis, G., Cardon, L. et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9, 356–369 (2008).
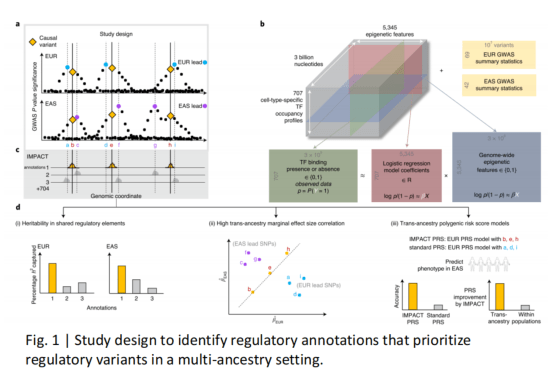
Amariuta, T. et al. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-typespecific regulatory elements. Nature Genetics 52, 1346–1354 (2020).
SNP A1 A2 freq b se p N
rs1001 A G 0.8493 0.0024 0.0055 0.6653 129850
rs1002 C G 0.0306 0.0034 0.0115 0.7659 129799
rs1003 A C 0.5128 0.0045 0.0038 0.2319 129830
...
# Select multiple associated SNPs through a stepwise selection procedure
gcta64 --bfile test --chr 1 --maf 0.01 --cojo-file test.ma --cojo-slct --out test_chr1
# Select a fixed number of of top associated SNPs through a stepwise selection procedure
gcta64 --bfile test --chr 1 --maf 0.01 --cojo-file test.ma --cojo-top-SNPs 10 --out test_chr1
# Estimate the joint effects of a subset of SNPs (given in the file test.snplist) without model selection
gcta64 --bfile test --chr 1 --extract test.snplist --cojo-file test.ma --cojo-joint --out test_chr1
# Perform single-SNP association analyses conditional on a set of SNPs (given in the file cond.snplist) without model selection
gcta64 --bfile test --chr 1 --maf 0.01 --cojo-file test.ma --cojo-cond cond.snplist --out test_chr1
输出文件格式
结尾为.jma的文件 (使用-cojo-slct 或 –cojo-joint时的输出)
Chr SNP bp freq refA b se p n freq_geno bJ bJ_se pJ LD_r
1 rs2001 172585028 0.6105 A 0.0377 0.0042 6.38e-19 121056 0.614 0.0379 0.0042 1.74e-19 -0.345
1 rs2002 174763990 0.4294 C 0.0287 0.0041 3.65e-12 124061 0.418 0.0289 0.0041 1.58e-12 0.012
1 rs2003 196696685 0.5863 T 0.0237 0.0042 1.38e-08 116314 0.589 0.0237 0.0042 1.67e-08 0.0
...
Chr SNP bp freq refA b se p n freq_geno bC bC_se pC
1 rs2001 172585028 0.6105 A 0.0377 0.0042 6.38e-19 121056 0.614 0.0379 0.0042 1.74e-19
1 rs2002 174763990 0.4294 C 0.0287 0.0041 3.65e-12 124061 0.418 0.0289 0.0041 1.58e-12
1 rs2003 196696685 0.5863 T 0.0237 0.0042 1.38e-08 116314 0.589 0.0237 0.0042 1.67e-08
...
GCTA-COJO 分析中参考样本的选择(重要!)
如果你的概括性数据来自单一的GWAS,那么最好的参考样本就是这个GWAS中的样本。
当概括性数据来自Meta分析,个体的基因型无法获取时,可以使用一个样本量较大队列的数据。例如,Wood et al. 2014 Nat Genet 使用了ARIC cohort (data available from dbGaP)
Conditional and joint analysis method: Yang et al. (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 44(4):369-375. [PubMed ID: 22426310]
GCTA software: Yang J, Lee SH, Goddard ME and Visscher PM. GCTA: a tool for Genome-wide Complex Trait Analysis. Am J Hum Genet. 2011 Jan 88(1): 76-82. [PubMed ID: 21167468]
./eagle -h
+-----------------------------+
| |
| Eagle v2.4.1 |
| November 18, 2018 |
| Po-Ru Loh |
| |
+-----------------------------+
Copyright (C) 2015-2018 Harvard University.
Distributed under the GNU GPLv3+ open source license.
Command line options:
eagle -h
Options:
--geneticMapFile arg HapMap genetic map provided with download:
tables/genetic_map_hg##.txt.gz
--outPrefix arg prefix for output files
--numThreads arg (=1) number of computational threads
Input options for phasing without a reference:
--bfile arg prefix of PLINK .fam, .bim, .bed files
--bfilegz arg prefix of PLINK .fam.gz, .bim.gz, .bed.gz
files
--fam arg PLINK .fam file (note: file names ending in
.gz are auto-decompressed)
--bim arg PLINK .bim file
--bed arg PLINK .bed file
--vcf arg [compressed] VCF/BCF file containing input
genotypes
--remove arg file(s) listing individuals to ignore (no
header; FID IID must be first two columns)
--exclude arg file(s) listing SNPs to ignore (no header;
SNP ID must be first column)
--maxMissingPerSnp arg (=0.1) QC filter: max missing rate per SNP
--maxMissingPerIndiv arg (=0.1) QC filter: max missing rate per person
Input/output options for phasing using a reference panel:
--vcfRef arg tabix-indexed [compressed] VCF/BCF file for
reference haplotypes
--vcfTarget arg tabix-indexed [compressed] VCF/BCF file for
target genotypes
--vcfOutFormat arg (=.) b|u|z|v: compressed BCF (b), uncomp BCF (u),
compressed VCF (z), uncomp VCF (v)
--noImpMissing disable imputation of missing target
genotypes (. or ./.)
--allowRefAltSwap allow swapping of REF/ALT in target vs. ref
VCF
--outputUnphased output unphased sites (target-only,
multi-allelic, etc.)
--keepMissingPloidyX assume missing genotypes have correct ploidy
(.=haploid, ./.=diploid)
--vcfExclude arg tabix-indexed [compressed] VCF/BCF file
containing variants to exclude from phasing
Region selection options:
--chrom arg (=0) chromosome to analyze (if input has many)
--bpStart arg (=0) minimum base pair position to analyze
--bpEnd arg (=1e9) maximum base pair position to analyze
--bpFlanking arg (=0) (ref-mode only) flanking region to use
during phasing but discard in output
Algorithm options:
--Kpbwt arg (=10000) number of conditioning haplotypes
--pbwtIters arg (=0) number of PBWT phasing iterations (0=auto)
--expectIBDcM arg (=2.0) expected length of haplotype copying (cM)
--histFactor arg (=0) history length multiplier (0=auto)
--genoErrProb arg (=0.003) estimated genotype error probability
--pbwtOnly in non-ref mode, use only PBWT iters
(automatic for sequence data)
--v1 use Eagle1 phasing algorithm (instead of
default Eagle2 algorithm)
[kaiwang@biocluster ~/]$ cat example/snplist.txt
rs74487784
rs41534544
rs4308095
rs12345678
[kaiwang@biocluster ~/]$ convert2annovar.pl -format rsid example/snplist.txt -dbsnpfile humandb/hg19_snp138.txt > snplist.avinput
NOTICE: Scanning dbSNP file humandb/hg19_snp138.txt...
NOTICE: input file contains 4 rs identifiers, output file contains information for 4 rs identifiers
WARNING: 1 rs identifiers have multiple records (due to multiple mapping) and they are all written to output
[kaiwang@biocluster ~/]$ cat snplist.avinput
chr2 186229004 186229004 C T rs4308095
chr7 6026775 6026775 T C rs41534544
chr7 6777183 6777183 G A rs41534544
chr9 3901666 3901666 T C rs12345678
chr22 24325095 24325095 A G rs74487784
chr2 186229004 186229004 C T
chr7 6026775 6026775 T C
chr7 6777183 6777183 G A
chr9 3901666 3901666 T C
chr22 24325095 24325095 A G
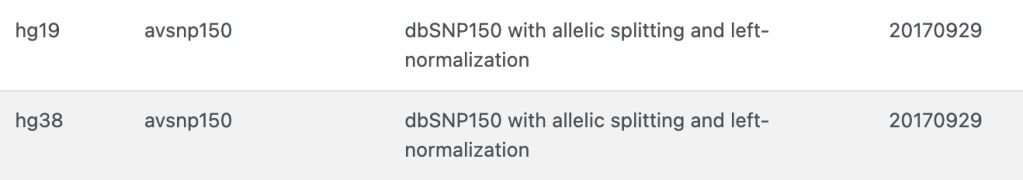
输出文件如下,ex1.avinput.hg19_avsnp150_dropped,
(注意某些SNP的rsID有合并等原因,版本不同rsID注释结果不一定相同)
avsnp150 rs4308095 chr2 186229004 186229004 C T
avsnp150 rs140195 chr22 24325095 24325095 A G
avsnp150 rs2228006 chr7 6026775 6026775 T C
avsnp150 rs766725467 chr7 6777183 6777183 G A
avsnp150 rs12345678 chr9 3901666 3901666 T C
#For Binary traits:
Rscript step1_fitNULLGLMM.R \
--plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly \
--phenoFile=./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt \
--phenoCol=y_binary \
--covarColList=x1,x2 \
--sampleIDColinphenoFile=IID \
--traitType=binary \
--outputPrefix=./output/example_binary \
--nThreads=4 \
--LOCO=FALSE \
--IsOverwriteVarianceRatioFile ## v0.38. Whether to overwrite the variance ratio file if the file already exists
#For Quantitative traits, if not normally distributed, inverse normalization needs to be specified to be TRUE --invNormalize=TRUE
Rscript step1_fitNULLGLMM.R \
--plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly \
--phenoFile=./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt \
--phenoCol=y_quantitative \
--covarColList=x1,x2 \
--sampleIDColinphenoFile=IID \
--traitType=quantitative \
--invNormalize=TRUE \
--outputPrefix=./output/example_quantitative \
--nThreads=4 \\
--LOCO=FALSE \\
--tauInit=1,0
共有三个输出文件:
.rda文件,包含空模型
30个随机选择的SNP的关联结果
包含估计的方差比值的文本文件 (回忆上述SAIGE算法中的r
.rda文件,包含空模型
./output/example_quantitative.rda
#open R
R
#load the model file in R
load("./output/example_quantitative.rda")
names(modglmm)
modglmm$theta
#theta: a vector of length 2. The first element is the dispersion parameter estimate and the second one is the variance component parameter estimate
#coefficients: fixed effect parameter estimates
#linear.predictors: a vector of length N (N is the sample size) containing linear predictors
#fitted.values: a vector of length N (N is the sample size) containing fitted mean values on the original scale
#Y: a vector of length N (N is the sample size) containing final working vector
#residuals: a vector of length N (N is the sample size) containing residuals on the original scale
#sampleID: a vector of length N (N is the sample size) containing sample IDs used to fit the null model
30个随机选择的SNP的关联结果
less -S ./output/example_quantitative_30markers.SAIGE.results.txt
方差比值文件 variance ratio file
less -S ./output/example_quantitative.varianceRatio.txt
CHR: chromosome
POS: genome position
SNPID: variant ID
Allele1: allele 1
Allele2: allele 2
AC_Allele2: allele count of allele 2
AF_Allele2: allele frequency of allele 2
imputationInfo: imputation info. If not in dosage/genotype input file, will output 1
N: sample size
BETA: effect size of allele 2
SE: standard error of BETA
Tstat: score statistic of allele 2
#SPA后的p值
p.value: p value (with SPA applied for binary traits)
p.value.NA: p value when SPA is not applied (only for binary traits)
Is.SPA.converge: whether SPA is converged or not (only for binary traits)
varT: estimated variance of score statistic with sample relatedness incorporated
varTstar: variance of score statistic without sample relatedness incorporated
AF.Cases: allele frequency of allele 2 in cases (only for binary traits and if --IsOutputAFinCaseCtrl=TRUE)
AF.Controls: allele frequency of allele 2 in controls (only for binary traits and if --IsOutputAFinCaseCtrl=TRUE)
Tstat_cond, p.value_cond, varT_cond, BETA_cond, SE_cond: summary stats for conditional analysis
Zhou, W. et al. (SAIGE)Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).