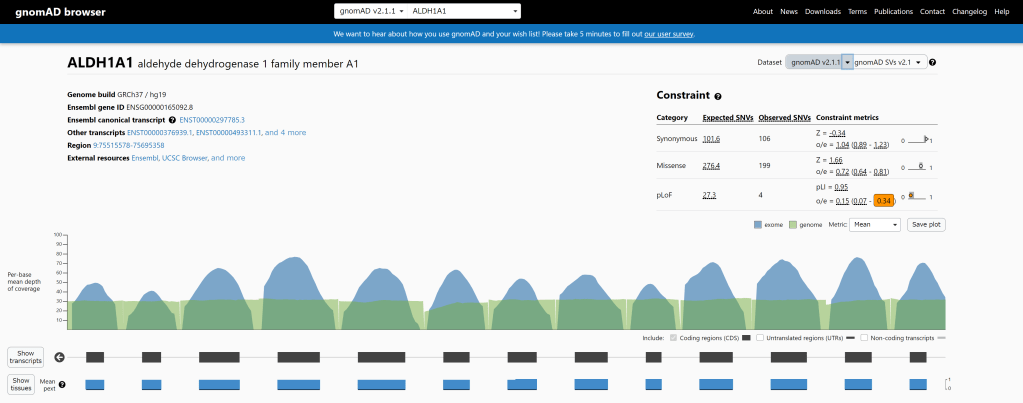
MR-MEGA 简介
MR-MEGA (Meta-Regression of Multi-Ethnic Genetic Association 多族裔遗传相关的荟萃回归) 是一款通过跨族裔荟萃回归来检测并精准定位复杂表型关联信号的工具。 该工具利用不同群体的全基因组概括性数据通过MDS方法推导遗传变异的轴。SNP等位的效应则根据其标准差的大小进行加权,加权后的效应就可以纳入线性回归的框架中,同时纳入遗传变异的轴作为协变量。该方法的灵活性使得我们可以将异质性分解为来自祖先以及残差这两个成分,也因此可以提高因果SNP精准定位的精度。
背景
此方法之前跨族裔荟萃分析中常使用的软件为同一研究组开发的MANTRA,相比于传统的固定以及随机效应荟萃分析,MANTRA在存在异质性时能够提高检验的power并提高跨族裔精确定位的精度。但由于MANTRA利用马尔科夫蒙特卡洛法来估计模型参数的后验分布,对于研究数较多的荟萃分析进行计算时,需要大量的计算资源。所以MANTRA的作者们又开发了MR-MEGA的方法,与MANTRA相比,MR-MEGA在保持power与精度的基础上,大幅削减了所需的计算资源。
方法核心原理
考虑对某一表型的 K 个GWAS研究,将第k个GWAS中的第j个SNP的参考等位频率表示为pkj,首先构建一个GWAS两两间欧几里得距离的矩阵,其中各项为由常染色体SNP计算得到,表示为D=[dkk’]。其中:
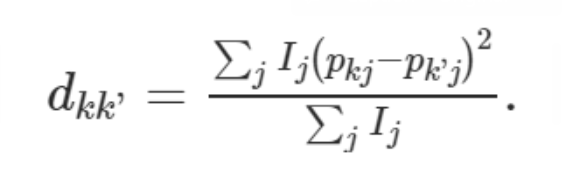
这个式子里,Ij为一个指示变量,表示第j个变异是否纳入距离计算之中。
接下来对这个距离矩阵D利用MDS(multi-dimensional scaling)方法进行降维,以得到T个遗传变异的轴,第k个GWAS的轴记为Xk。注意,这里遗传变异的轴的个数的选择应当基于GWAS中群体多样性,限制为T≤K-2。
对于第k个GWAS中的第j个变异,分别用bkj与vkj来表示参考等位的效应大小与其方差。于是我们就可以建立一个关于参考等位的效应的线性回归方程,
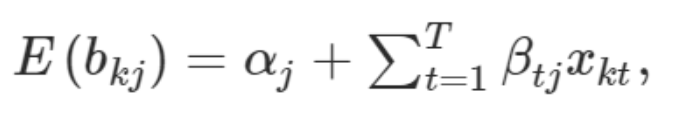
其中αj是截距,βtj是第j个SNP在第t个轴上的效应。第k个GWAS贡献则通过第j个SNP的倒方差来计算权重,记为v-1kj。于是我们可以将截距α视为各个轴上0所表示的群体中第j个SNP的期望效应值。
以此我们就能得到汇聚后的效应值,以及异质性检验的结果。
使用方法:
下载页面: https://genomics.ut.ee/en/tools/mr-mega
linux系统中下载完成后解压,
unzip MRMEGA_v*.zip #解压
make #编译
./MR-MEGA #测试是否安装成功
make后将MR-MEGA添加进环境(可选)
输入文件:
输入文件 1. 首先是各个GWAS的概括性数据,格式要求如下
每个GWAS的概括性数据都必须有以下的列,并由空格或分隔符分隔:
1) MARKERNAME – snp name
2) EA – effect allele
3) NEA – non effect allele
4) OR - odds ratio
5) OR_95L - lower confidence interval of OR
6) OR_95U - upper confidence interval of OR
7) EAF – effect allele frequency
8) N - sample size
9) CHROMOSOME - chromosome of marker
10) POSITION - position of marker
当表型是数量型表型时,使用beta:
- BETA – beta
- SE – std. error
以下的列可选,默认是所有的SNP都在正链上:
- STRAND – marker strand (if the column is missing then program expects all markers being on positive strand)
例如:
MARKERNAME STRAND CHROMOSOME POSITION IMP EA NEA EAF N BETA SE
rs12565286 + 1 761153 0 G C 0.3 1200 -0.02 0.0403
rs2977670 + 1 763754 0 C G 0.23 1200 -0.01 0.40612
rs12138618 + 1 790098 0 G A 0.97 1200 -0.07 0.37
rs3094315 + 1 792429 0 G A 0.01 1199 0.0258 0.1012
rs3131968 + 1 794055 0 G A 0.27 1200 -0.373 0.0101
rs2519016 + 1 805811 0 T C 0.04 1200 0.26 0.3472
rs12562034 + 1 808311 0 G A 0.65 1200 0.0092 0.2
输入文件 2, 除此之外我们还要准备一个存储GWAS概括性数据文件名的列表,(默认名称为 mr-mega.in)
Pop1.txt.gz
Pop2.txt.gz
Pop3.txt.gz
Pop4.txt.gz
Pop5.txt.gz
Pop6.txt.gz
Pop7.txt.gz
Pop8.txt.gz
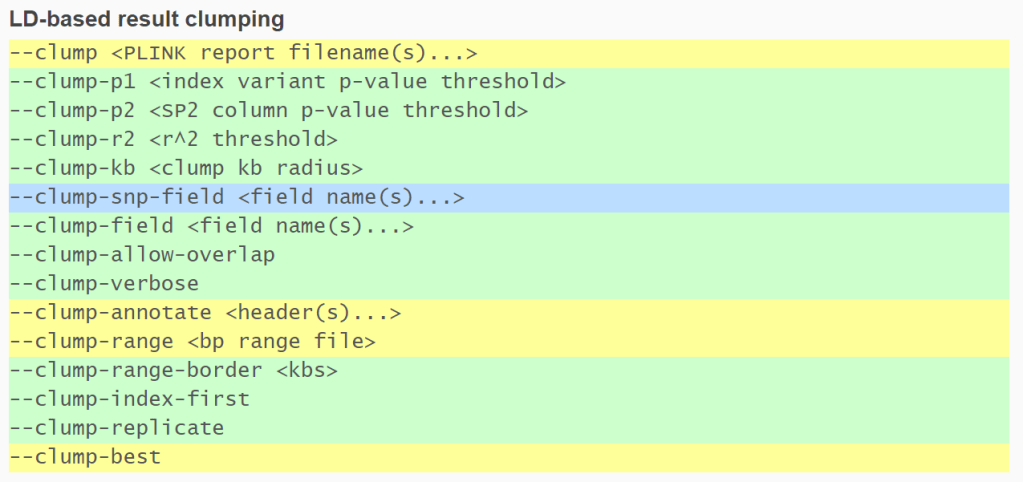
MR-MEGA基本语法
./MR-MEGA [--name_pos <string>] ... [--name_chr <string>] ...
[--name_n <string>] ... [--name_strand <string>] ...
[--name_or_95u <string>] ... [--name_or_95l <string>] ...
[--name_or <string>] ... [--name_se <string>] ...
[--name_beta <string>] ... [--name_eaf <string>] ...
[--name_nea <string>] ... [--name_ea <string>] ...
[--name_marker <string>] ... [-f <string>] ... [--pc <int>]
[-t <double>] [--no_std_names] [--debug] [--qt] [--gco]
[--gc] [--no_alleles] [-m <string>] [-o <string>] [-i
<string>] [--] [--version] [-h]
—name_xxx都是用来制定自定义列名
-f 可以对列进行过滤 例如 INFO>0.4
-pc <int>指定纳入回归的pc数量,默认为4,但要注意,PC数必须小于研究数-2,如果有5个研究,那么能够纳入PC的最大数量是2.
-qt 数量表型
-o <string> 指定输出前缀
-i <string> 制定输入的文件列表
例:
./MR-MEGA -i mr-mega.in -o mr-mega-out —pc 2
结果解读
MR-MEGA成功运行后会生成两个后缀分别为*.result 和 *.log的文件:
result文件中包含以下列:
#基本信息
MarkerName - unique marker identification across input files
Chromosome - chromosome of marker
Position - physical position in chromosome of marker
EA - allele, which effect was measured across input files
NEA - other allele
EAF - average effect allele frequency (weighted by the samplesize of each input file)
Nsample - total number of samples
Ncohort - total number of cohorts, where the marker was present
#效应
Effects - effect direction across cohorts (+ if the effect allele effect was positive, - if negative, 0 if the effect was zero, ? if marker was not available in cohort)
beta_0 - effect of first PC of meta-regression
se_0 - stderr of the effect of first PC of meta-regression
(beta_1)
(se_1)
(...)
#统计量与p值
chisq_association - chisq value of the association
ndf_association - number of degrees of freedom of the association
P-value_association - p-value of the association
#异质性与p值
chisq_ancestry_het - chisq value of the heterogeneity due to different ancestry
ndf_ancestry_het - ndf of the heterogeneity due to different ancestry
P-value_ancestry_het - p-value of the heterogeneity due to different ancestry
chisq_residual_het - chisq value of the residual heterogeneity
ndf_residual_het - ndf of the residual heterogeneity
P-value_residual_het - p-value of the residual heterogeneity
lnBF - log of Bayes factor
Comments - reason why marker was not analysed
参考:
https://genomics.ut.ee/en/tools/mr-mega
Reedik Mägi, Momoko Horikoshi, Tamar Sofer, Anubha Mahajan, Hidetoshi Kitajima, Nora Franceschini, Mark I. McCarthy, COGENT-Kidney Consortium, T2D-GENES Consortium, Andrew P. Morris, Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution, Human Molecular Genetics, Volume 26, Issue 18, 15 September 2017, Pages 3639–3650, https://doi.org/10.1093/hmg/ddx280
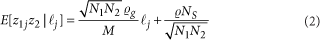







 是某个等位基因在整个群体里的频率,
是某个等位基因在整个群体里的频率,  是等位基因在不同亚群体之间的被群体大小加权后的频率的方差(组间方差),
是等位基因在不同亚群体之间的被群体大小加权后的频率的方差(组间方差), 是整个群体的等位基因频率的方差。那么Fst可以被定义为:
是整个群体的等位基因频率的方差。那么Fst可以被定义为:


 则是 给定两个来自总体的个体,这两个个体血缘同源(IBD)的概率。
则是 给定两个来自总体的个体,这两个个体血缘同源(IBD)的概率。 
 与
与 分别代表两个不同亚群或相同亚群的个体之间,成对等位基因之间不同的平均值(average number of pairwise differences)。
分别代表两个不同亚群或相同亚群的个体之间,成对等位基因之间不同的平均值(average number of pairwise differences)。