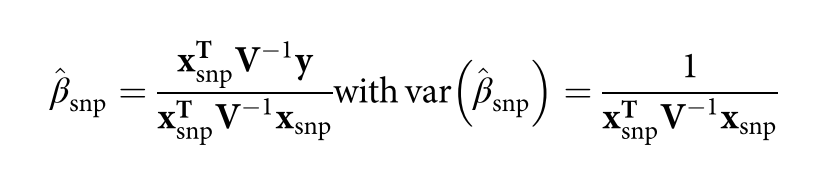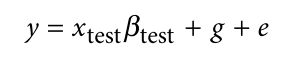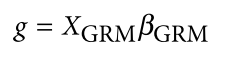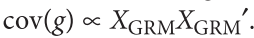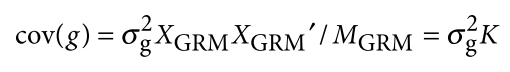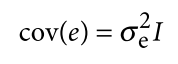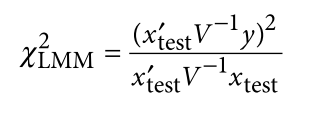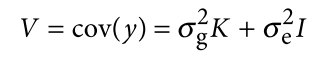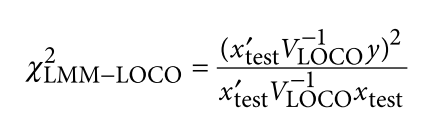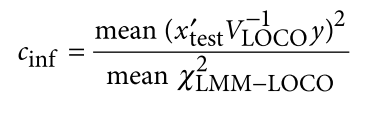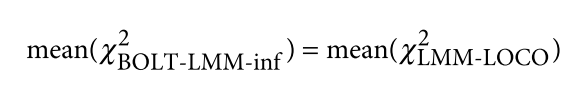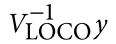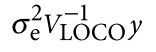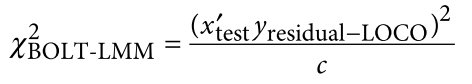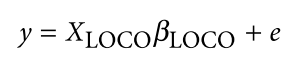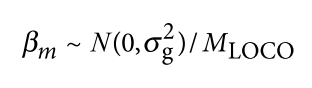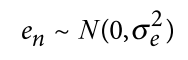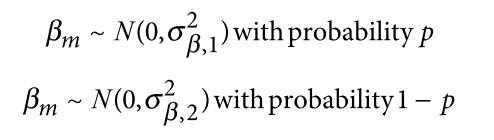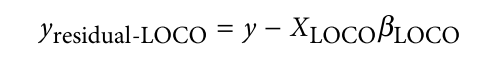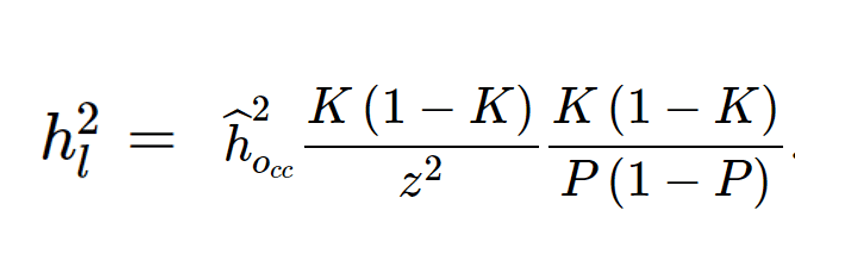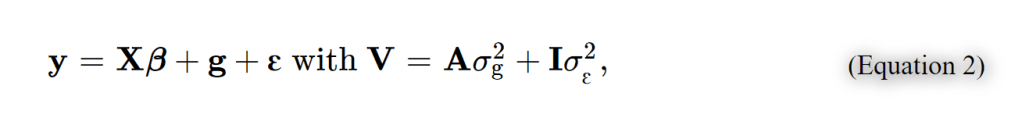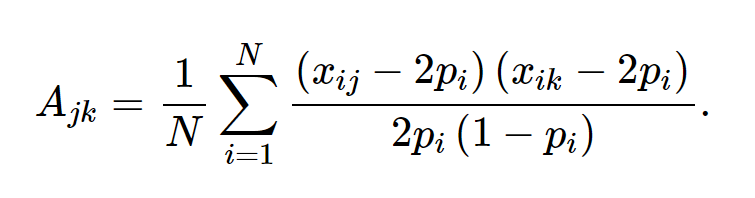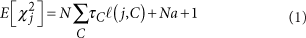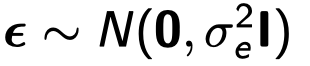Jiang L, Zheng Z, Qi T, et al. A resource-efficient tool for mixed model association analysis of large-scale data[J]. Nature genetics, 2019, 51(12): 1749-1755.
Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88(3):294-305. doi:10.1016/j.ajhg.2011.02.002
Lee SH 等人的文章介绍了转换的方法,当case和control并不是从人群中随机抽取的时候,转换公式如下:
hl2为 易感性尺度的遗传力 ,ho2为 观测尺度遗传力
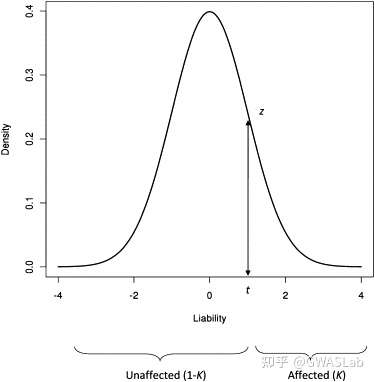
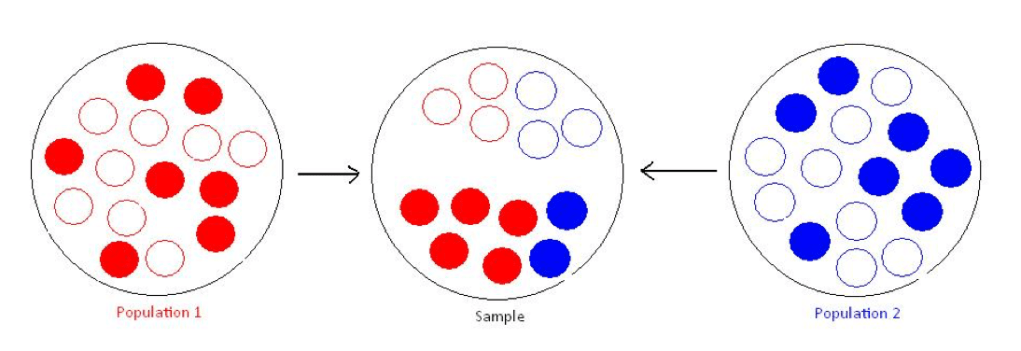
K为人群中病例的比例(流行率)
P为抽取的样本中病例的比例
z则为正态分布的密度函数的在所取阈值t处的值
使用R进行转换
#K = pop prevalence 人群中流行率
#P = proportion of cases in study 样本中病例比例
#h2 = Heritability estimate (on observed scale) 观测尺度遗传力
#T = liability threshold 易感性阈值
#zv = 正态分布的密度函数的在所取阈值t处的值
K=0.0659
P=0.0659
h2=0.0365
zv <- dnorm(qnorm(K))
h2_liab <- h2 * K^2 * ( 1 - K)^2 / P / (1-P) / zv^2
h2_liab
该方法的作者们基于多个公开的注释数据库,构建了不针对任何细胞类型的全基线模型 ‘full baseline model’ , 包括 coding, UTR, promoter and intronic regions the histone marks monomethylation (H3K4me1) and trimethylation (H3K4me3) of histone H3 at lysine 等等。除此之外,还基于 全基线模型 ,构建了多个针对特定细胞类型的模型,包含针对细胞类型的注释等。
为了避免此现象造成的power损失,理论上在构建null模型中排除掉待检验SNP是正确的做法,但这样太占运算资源,所以在实践中,我们会采用 LOCO Leave-one-chromosome-out ,即使用排除掉待检验SNP所在的染色体的所有SNP,再进行检验(也就是说我们有对应22个常染色体的loco null模型)。