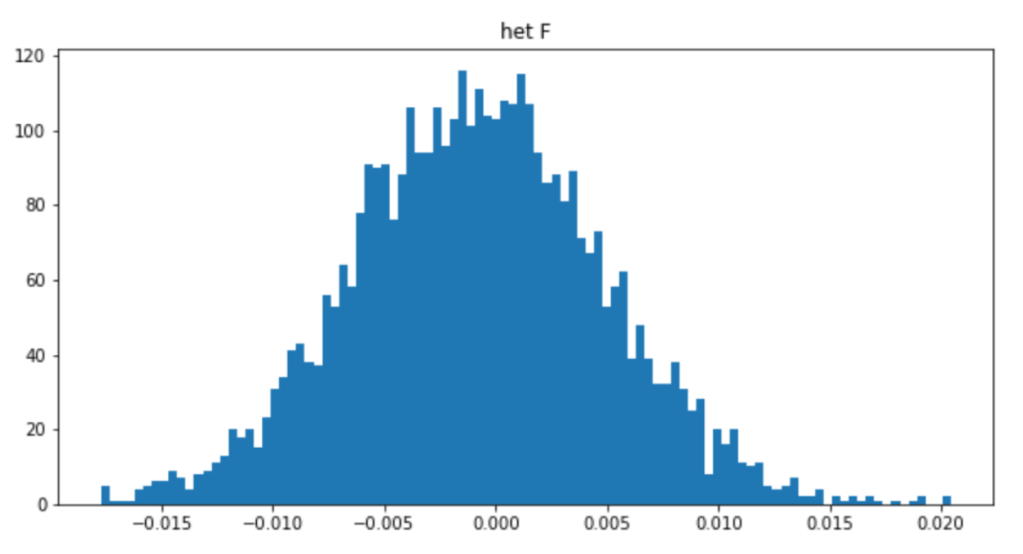
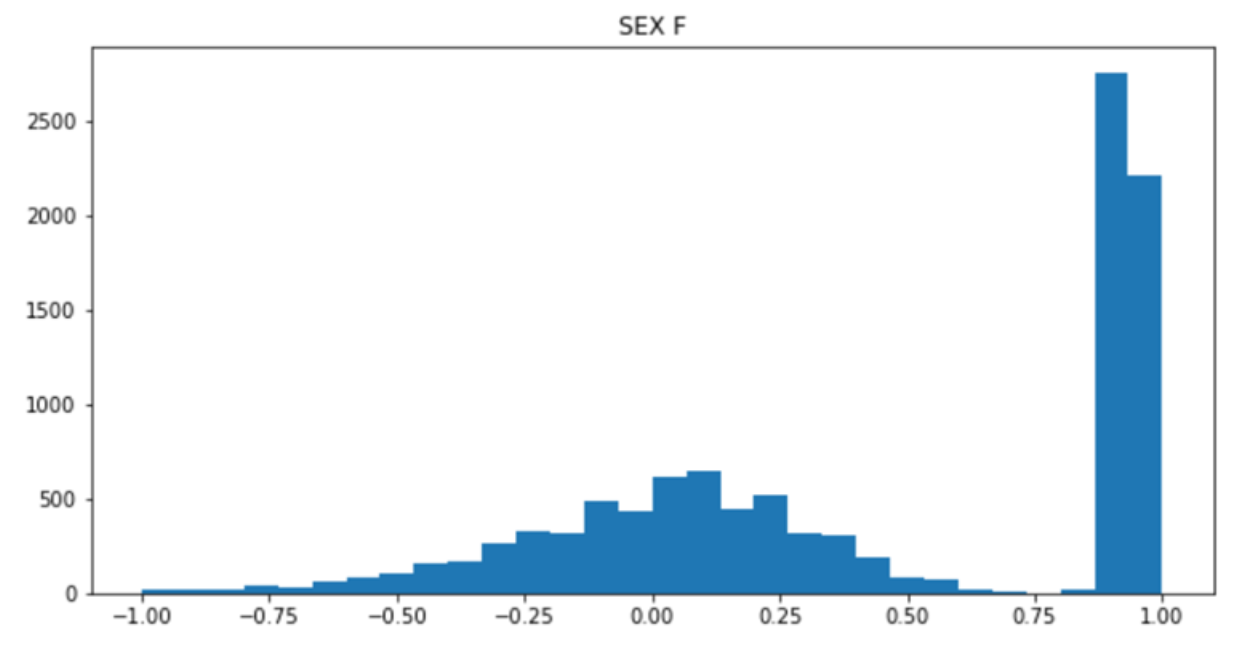
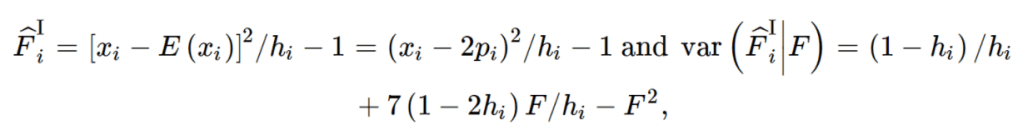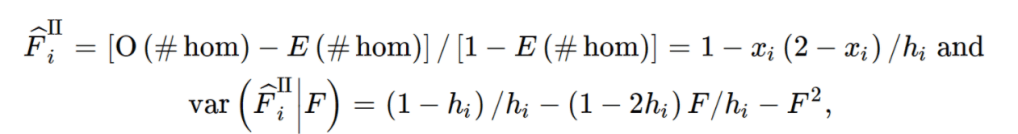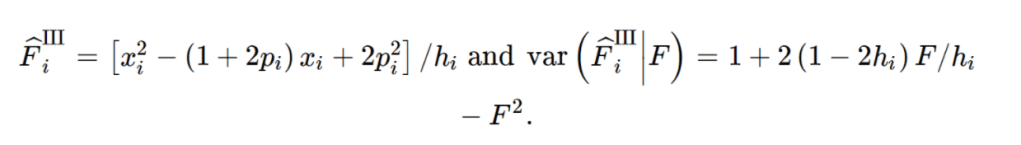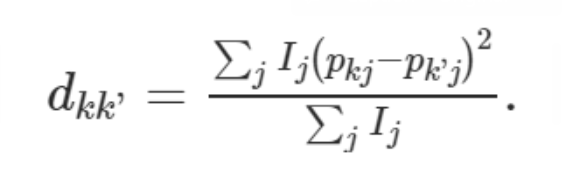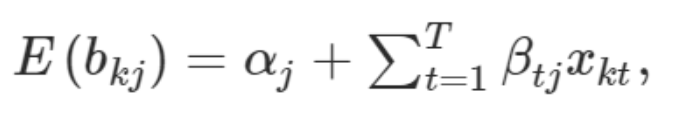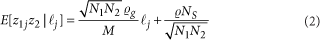import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
het = pd.read_csv("gwa.het","\\s+")
plt.figure(figsize=(10,5))
plt.hist(het["F"],bins=100)
plt.title("het F")
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to HapMap_3_r3_1.log.
Options in effect:
--bfile HapMap_3_r3_1
--check-sex
--extract HapMap_3_r3_1.prune.in
--out HapMap_3_r3_1
191875 MB RAM detected; reserving 95937 MB for main workspace.
Allocated 3037 MB successfully, after larger attempt(s) failed.
1457897 variants loaded from .bim file.
165 people (80 males, 85 females) loaded from .fam.
112 phenotype values loaded from .fam.
--extract: 166240 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 112 founders and 53 nonfounders present.
Calculating allele frequencies... done.
Warning: 35 het. haploid genotypes present (see HapMap_3_r3_1.hh ); many
commands treat these as missing.
Total genotyping rate is 0.996691.
166240 variants and 165 people pass filters and QC.
Among remaining phenotypes, 56 are cases and 56 are controls. (53 phenotypes
are missing.)
--check-sex: 3993 Xchr and 0 Ychr variant(s) scanned, 1 problem detected.
Report written to HapMap_3_r3_1.sexcheck .
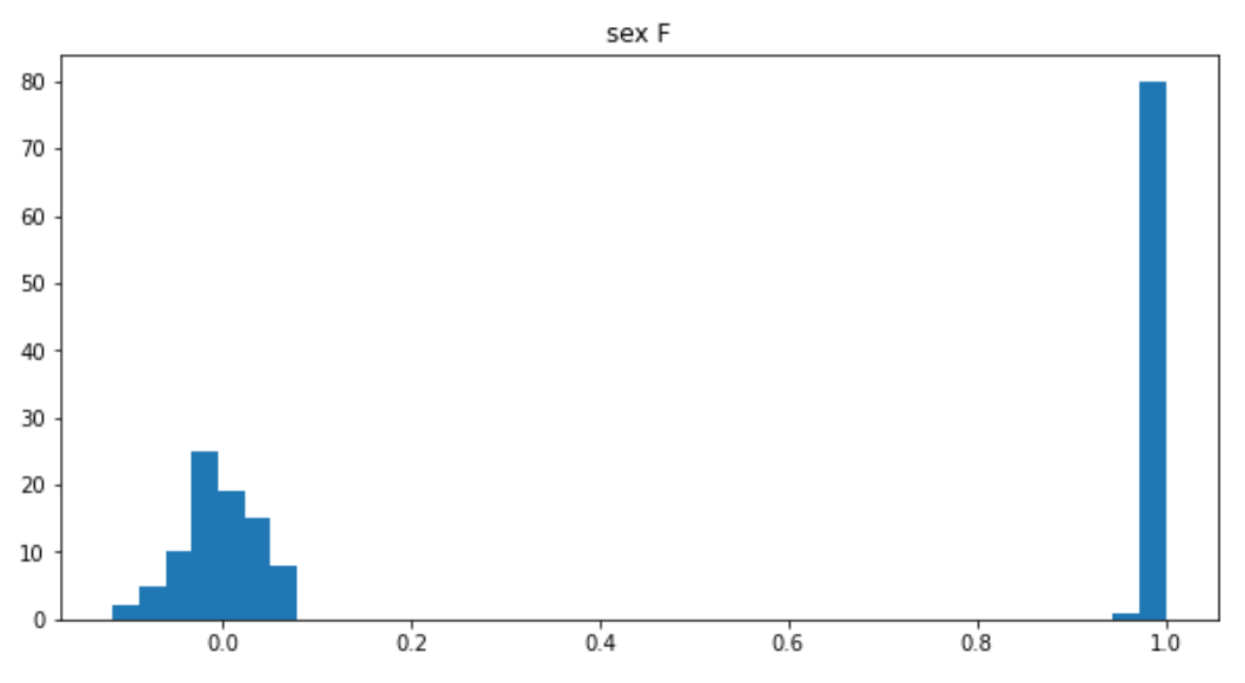
–check-sex: 3993 Xchr and 0 Ychr variant(s) scanned, 1 problem detected.从日志文件中可以看出我们检测出了1个不一致的样本。上述代码运行结果如下:
head HapMap_3_r3_1.sexcheck
FID IID PEDSEX SNPSEX STATUS F
1328 NA06989 2 2 OK 0.02151
1377 NA11891 1 1 OK 1
1349 NA11843 1 1 OK 1
1330 NA12341 2 2 OK 0.009813
1444 NA12739 1 1 OK 1
1344 NA10850 2 2 OK 0.0533
1328 NA06984 1 1 OK 0.9989
1463 NA12877 1 1 OK 1
1418 NA12275 2 2 OK -0.0747
孟德尔随机化的核心其实是利用了孟德尔第二定律,也就是自由组合规律(law of independent assortment),当具有两对(或更多对)相对性状的亲本进行杂交,在子一代产生配子时,在等位基因分离的同时,非同源染色体上的基因表现为自由组合,这一过程类似于随机对照试验中的随机分组,所以我个人理解的孟德尔随机化就是 基于孟德尔第二定律的随机对照试验。
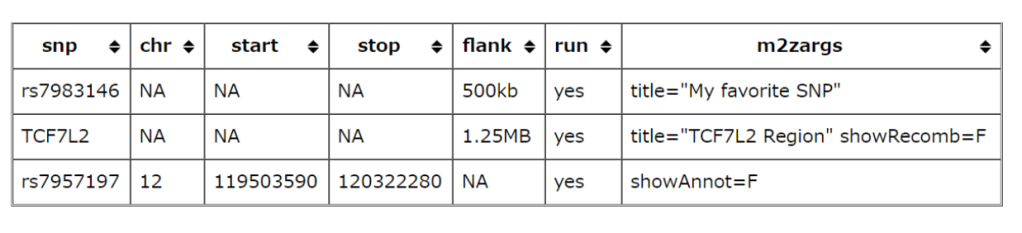
cd <directory where you want to place locuszoom>
tar zxf /path/to/locuszoom.tgz
ln -s bin/locuszoom /usr/local/bin/locuszoom #根据自己的环境变量制定
locuszoom的文件结构主要包括的内容如下所示:
locuszoom/
bin/
locuszoom (this is the locuszoom "executable")
locuszoom.R (the R script which is used by locuszoom for creating the plots)
dbmeister.py (script for creating custom user databases)
lzupdate.py (script for creating an updated copy of the provided locuszoom database)
conf/ (configuration file located here)
data/
database/ (SQLite file located here)
hapmap/ (hapmap genotype files)
1000G/ (1000G genotype files)
src/ (source code for locuszoom)
MR-MEGA (Meta-Regression of Multi-Ethnic Genetic Association 多族裔遗传相关的荟萃回归) 是一款通过跨族裔荟萃回归来检测并精准定位复杂表型关联信号的工具。 该工具利用不同群体的全基因组概括性数据通过MDS方法推导遗传变异的轴。SNP等位的效应则根据其标准差的大小进行加权,加权后的效应就可以纳入线性回归的框架中,同时纳入遗传变异的轴作为协变量。该方法的灵活性使得我们可以将异质性分解为来自祖先以及残差这两个成分,也因此可以提高因果SNP精准定位的精度。
unzip MRMEGA_v*.zip #解压
make #编译
./MR-MEGA #测试是否安装成功
make后将MR-MEGA添加进环境(可选)
输入文件:
输入文件 1. 首先是各个GWAS的概括性数据,格式要求如下
每个GWAS的概括性数据都必须有以下的列,并由空格或分隔符分隔:
1) MARKERNAME – snp name
2) EA – effect allele
3) NEA – non effect allele
4) OR - odds ratio
5) OR_95L - lower confidence interval of OR
6) OR_95U - upper confidence interval of OR
7) EAF – effect allele frequency
8) N - sample size
9) CHROMOSOME - chromosome of marker
10) POSITION - position of marker
当表型是数量型表型时,使用beta:
BETA – beta
SE – std. error
以下的列可选,默认是所有的SNP都在正链上:
STRAND – marker strand (if the column is missing then program expects all markers being on positive strand)
例如:
MARKERNAME STRAND CHROMOSOME POSITION IMP EA NEA EAF N BETA SE
rs12565286 + 1 761153 0 G C 0.3 1200 -0.02 0.0403
rs2977670 + 1 763754 0 C G 0.23 1200 -0.01 0.40612
rs12138618 + 1 790098 0 G A 0.97 1200 -0.07 0.37
rs3094315 + 1 792429 0 G A 0.01 1199 0.0258 0.1012
rs3131968 + 1 794055 0 G A 0.27 1200 -0.373 0.0101
rs2519016 + 1 805811 0 T C 0.04 1200 0.26 0.3472
rs12562034 + 1 808311 0 G A 0.65 1200 0.0092 0.2
#基本信息
MarkerName - unique marker identification across input files
Chromosome - chromosome of marker
Position - physical position in chromosome of marker
EA - allele, which effect was measured across input files
NEA - other allele
EAF - average effect allele frequency (weighted by the samplesize of each input file)
Nsample - total number of samples
Ncohort - total number of cohorts, where the marker was present
#效应
Effects - effect direction across cohorts (+ if the effect allele effect was positive, - if negative, 0 if the effect was zero, ? if marker was not available in cohort)
beta_0 - effect of first PC of meta-regression
se_0 - stderr of the effect of first PC of meta-regression
(beta_1)
(se_1)
(...)
#统计量与p值
chisq_association - chisq value of the association
ndf_association - number of degrees of freedom of the association
P-value_association - p-value of the association
#异质性与p值
chisq_ancestry_het - chisq value of the heterogeneity due to different ancestry
ndf_ancestry_het - ndf of the heterogeneity due to different ancestry
P-value_ancestry_het - p-value of the heterogeneity due to different ancestry
chisq_residual_het - chisq value of the residual heterogeneity
ndf_residual_het - ndf of the residual heterogeneity
P-value_residual_het - p-value of the residual heterogeneity
lnBF - log of Bayes factor
Comments - reason why marker was not analysed
Reedik Mägi, Momoko Horikoshi, Tamar Sofer, Anubha Mahajan, Hidetoshi Kitajima, Nora Franceschini, Mark I. McCarthy, COGENT-Kidney Consortium, T2D-GENES Consortium, Andrew P. Morris, Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution, Human Molecular Genetics, Volume 26, Issue 18, 15 September 2017, Pages 3639–3650, https://doi.org/10.1093/hmg/ddx280
Bulik-Sullivan, B., Finucane, H., Anttila, V. et al. An atlas of genetic correlations across human diseases and traits. Nat Genet47, 1236–1241 (2015). https://doi.org/10.1038/ng.3406
Bulik-Sullivan, B., Finucane, H., Anttila, V. et al. An atlas of genetic correlations across human diseases and traits. Nat Genet47, 1236–1241 (2015). https://doi.org/10.1038/ng.3406
1.Timpson, N. J., Greenwood, C. M. T., Soranzo, N., Lawson, D. J. & Richards, J. B. Genetic architecture: The shape of the genetic contribution to human traits and disease. Nature Reviews Genetics19, 110–124 (2018).
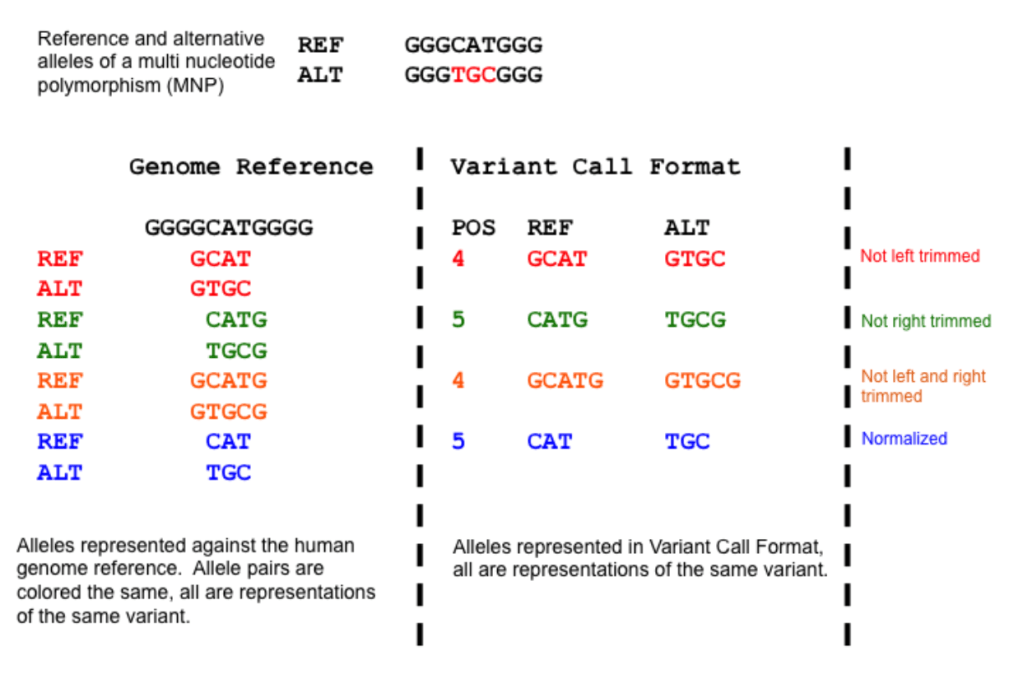
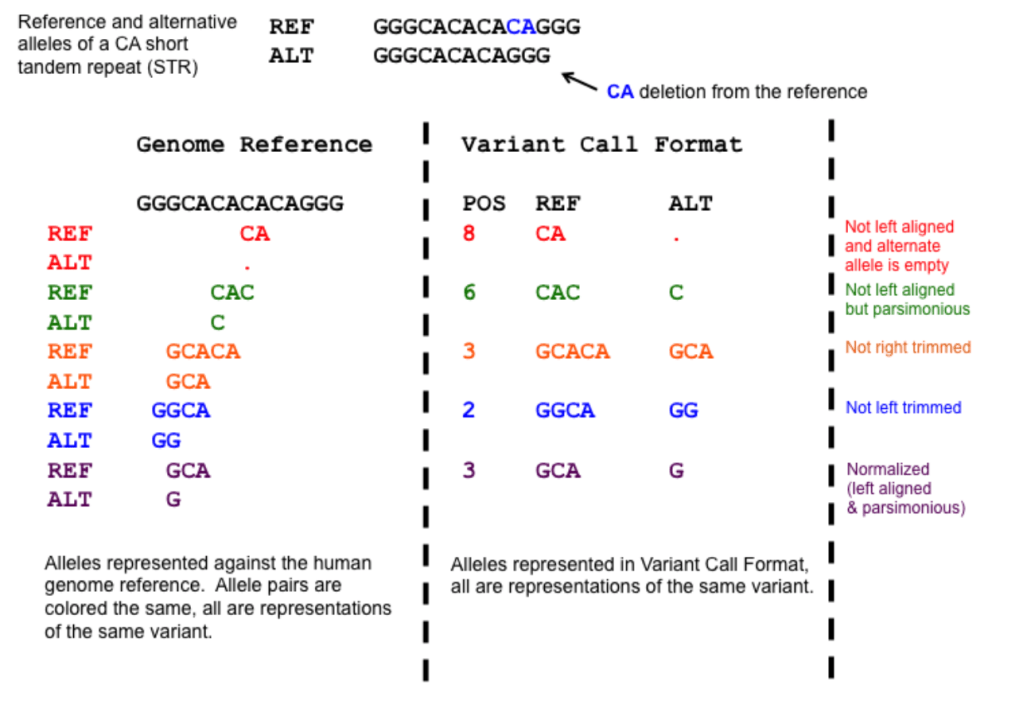
-protocol refGene,cytoBand,exac03,avsnp147,dbnsfp30a : 所用的数据库包括 ExAC version 0.3 (exac03) dbNFSP version 3.0a (dbnsfp30a), dbSNP version 147 with left-normalization (avsnp147) 数据库
-operation gx,r,f,f,f :指定针对protocol的操作(与-protocol一一对应),可选的操作包括g (gene-based), gx (gene-based with cross-reference annotation from -xref argument), r (region-based) 以及 f(filter-based).
第一至五列为必须,分别是染色体号,起始位点,结束位点,参考等位基因 reference allele 以及 替代等位基因 alternative allele,第五列之后可自由添加所需要的信息
1 948921 948921 T C comments: rs15842, a SNP in 5' UTR of ISG15
1 1404001 1404001 G T comments: rs149123833, a SNP in 3' UTR of ATAD3C
1 5935162 5935162 A T comments: rs1287637, a splice site variant in NPHP4
1 162736463 162736463 C T comments: rs1000050, a SNP in Illumina SNP arrays
1 84875173 84875173 C T comments: rs6576700 or SNP_A-1780419, a SNP in Affymetrix SNP arrays
1 13211293 13211294 TC - comments: rs59770105, a 2-bp deletion
1 11403596 11403596 - AT comments: rs35561142, a 2-bp insertion
1 105492231 105492231 A ATAAA comments: rs10552169, a block substitution
1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn's disease
2 234183368 234183368 A G comments: rs2241880 (T300A), a SNP in the ATG16L1 associated with Crohn's disease
16 50745926 50745926 C T comments: rs2066844 (R702W), a non-synonymous SNP in NOD2
16 50756540 50756540 G C comments: rs2066845 (G908R), a non-synonymous SNP in NOD2
16 50763778 50763778 - C comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2
13 20763686 20763686 G - comments: rs1801002 (del35G), a frameshift mutation in GJB2, associated with hearing loss
13 20797176 21105944 0 - comments: a 342kb deletion encompassing GJB6, associated with hearing loss
annotate_variation.pl -downdb -buildver hg19 -webfrom annovar refGene humandb/
NOTICE: Web-based checking to see whether ANNOVAR new version is available ... Done
NOTICE: Downloading annotation database http://www.openbioinformatics.org/annovar/download/hg19_refGene.txt.gz ... OK
NOTICE: Downloading annotation database http://www.openbioinformatics.org/annovar/download/hg19_refLink.txt.gz ... OK
NOTICE: Downloading annotation database http://www.openbioinformatics.org/annovar/download/hg19_refGeneMrna.fa.gz ... OK
NOTICE: Uncompressing downloaded files
NOTICE: Finished downloading annotation files for hg19 build version, with files saved at the 'humandb' directory
annotate_variation.pl -out ex1 -build hg19 example/ex1.avinput humandb/
NOTICE: The --geneanno operation is set to ON by default
NOTICE: Reading gene annotation from humandb/hg19_refGene.txt ... Done with 48660 transcripts (including 10375 without coding sequence annotation) for 25588 unique genes
NOTICE: Reading FASTA sequences from humandb/hg19_refGeneMrna.fa ... Done with 14 sequences
WARNING: A total of 333 sequences will be ignored due to lack of correct ORF annotation
NOTICE: Finished gene-based annotation on 15 genetic variants in example/ex1.avinput
NOTICE: Output files were written to ex1.variant_function, ex1.exonic_variant_function
cat ex1.variant_function
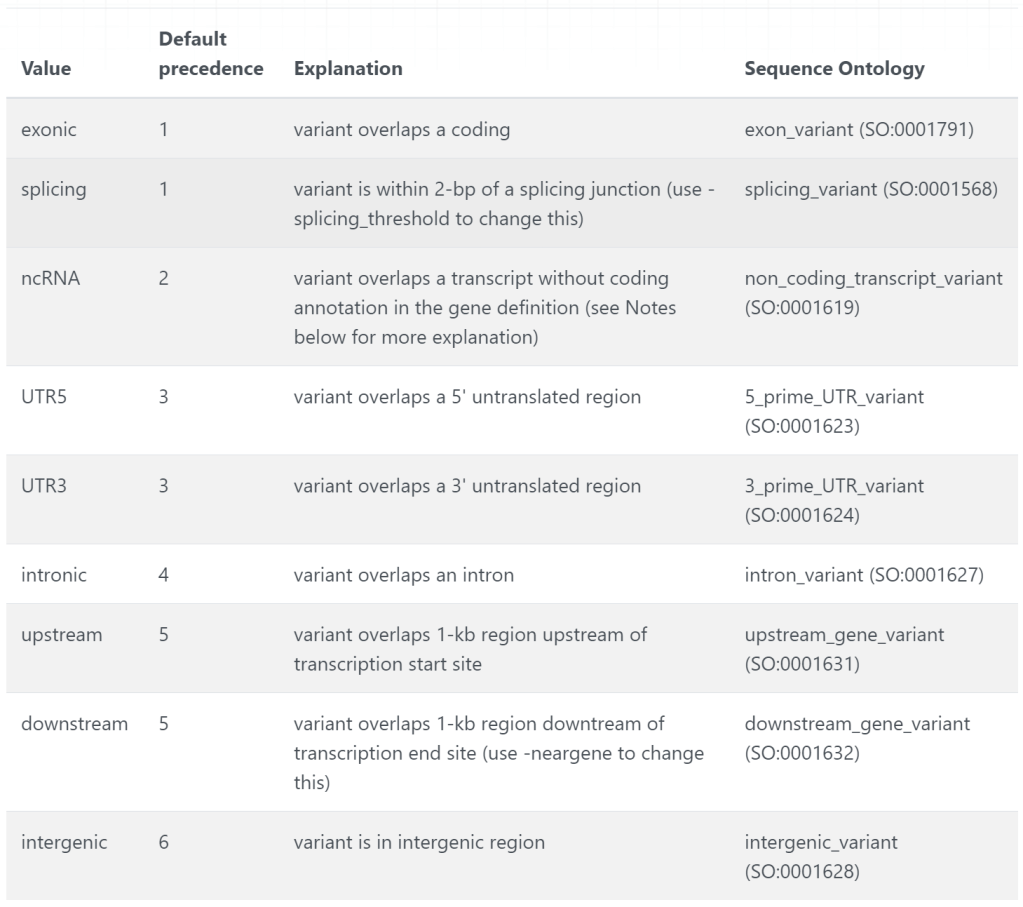
UTR5 ISG15(NM_005101:c.-33T>C) 1 948921 948921 T C comments: rs15842, a SNP in 5' UTR of ISG15
UTR3 ATAD3C(NM_001039211:c.*91G>T) 1 1404001 1404001 G T comments: rs149123833, a SNP in 3' UTR of ATAD3C
splicing NPHP4(NM_001291593:exon19:c.1279-2T>A,NM_001291594:exon18:c.1282-2T>A,NM_015102:exon22:c.2818-2T>A) 1 5935162 5935162 A T comments: rs1287637, a splice site variant in NPHP4
intronic DDR2 1 162736463 162736463 C T comments: rs1000050, a SNP in Illumina SNP arrays
intronic DNASE2B 1 84875173 84875173 C T comments: rs6576700 or SNP_A-1780419, a SNP in Affymetrix SNP arrays
intergenic LOC645354(dist=11566),LOC391003(dist=116902) 1 13211293 13211294 TC - comments: rs59770105, a 2-bp deletion
intergenic UBIAD1(dist=55105),PTCHD2(dist=135699) 1 11403596 11403596 - AT comments: rs35561142, a 2-bp insertion
intergenic LOC100129138(dist=872538),NONE(dist=NONE) 1 105492231 105492231 A ATAAA comments: rs10552169, a block substitution
exonic IL23R 1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn's disease
exonic ATG16L1 2 234183368 234183368 A G comments: rs2241880 (T300A), a SNP in the ATG16L1 associated with Crohn's disease
exonic NOD2 16 50745926 50745926 C T comments: rs2066844 (R702W), a non-synonymous SNP in NOD2
exonic NOD2 16 50756540 50756540 G C comments: rs2066845 (G908R), a non-synonymous SNP in NOD2
exonic NOD2 16 50763778 50763778 - C comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2
exonic GJB2 13 20763686 20763686 G - comments: rs1801002 (del35G), a frameshift mutation in GJB2, associated with hearing loss
exonic CRYL1,GJB6 13 20797176 21105944 0 - comments: a 342kb deletion encompassing GJB6, associated with hearing loss
cat ex1.exonic_variant_function
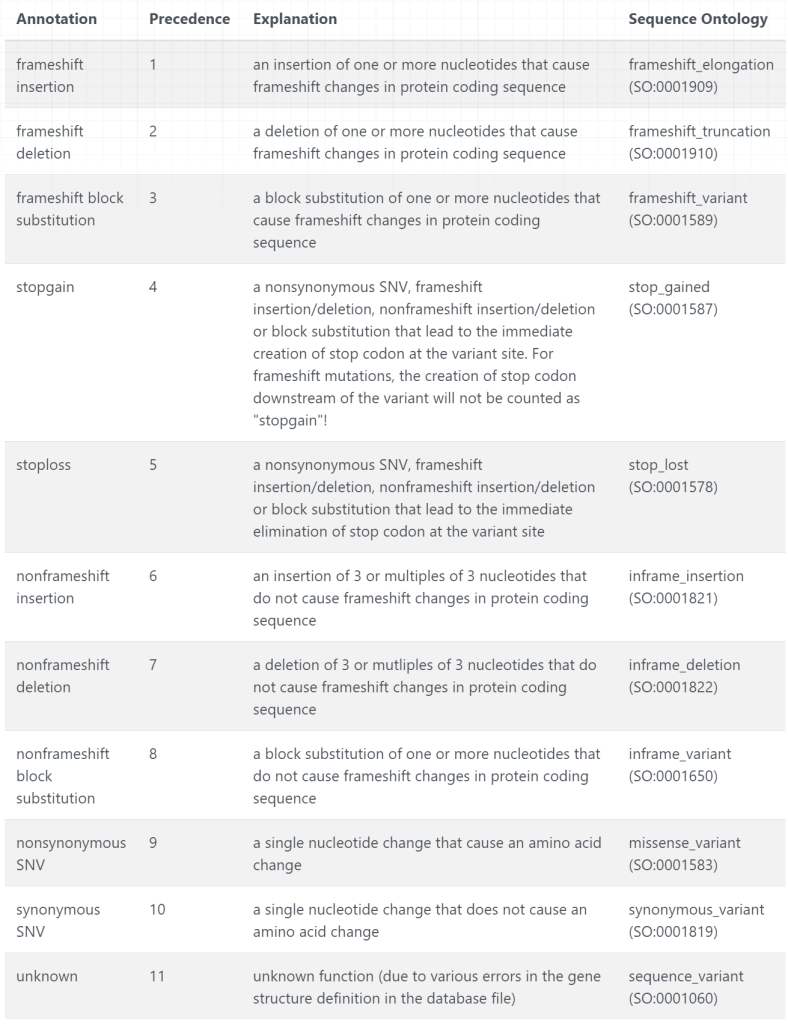
line9 nonsynonymous SNV IL23R:NM_144701:exon9:c.G1142A:p.R381Q, 1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn's disease
line10 nonsynonymous SNV ATG16L1:NM_001190267:exon9:c.A550G:p.T184A,ATG16L1:NM_017974:exon8:c.A841G:p.T281A,ATG16L1:NM_001190266:exon9:c.A646G:p.T216A,ATG16L1:NM_030803:exon9:c.A898G:p.T300A,ATG16L1:NM_198890:exon5:c.A409G:p.T137A, 2 234183368 234183368 A G comments: rs2241880 (T300A), a SNP in the ATG16L1 associated with Crohn's disease
line11 nonsynonymous SNV NOD2:NM_022162:exon4:c.C2104T:p.R702W,NOD2:NM_001293557:exon3:c.C2023T:p.R675W, 16 50745926 50745926 C comments: rs2066844 (R702W), a non-synonymous SNP in NOD2
line12 nonsynonymous SNV NOD2:NM_022162:exon8:c.G2722C:p.G908R,NOD2:NM_001293557:exon7:c.G2641C:p.G881R, 16 50756540 50756540 G comments: rs2066845 (G908R), a non-synonymous SNP in NOD2
line13 frameshift insertion NOD2:NM_022162:exon11:c.3017dupC:p.A1006fs,NOD2:NM_001293557:exon10:c.2936dupC:p.A979fs, 16 50763778 5076377comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2
line14 frameshift deletion GJB2:NM_004004:exon2:c.35delG:p.G12fs, 13 20763686 20763686 G - comments: rs1801002 (del35G), a frameshift mutation in GJB2, associated with hearing loss
line15 frameshift deletion GJB6:NM_001110221:wholegene,GJB6:NM_001110220:wholegene,GJB6:NM_001110219:wholegene,CRYL1:NM_015974:wholegene,GJB6:NM_006783:wholegene, 13 20797176 21105944 0 - comments: a 342kb deletion encompassing GJB6, associated with hearing loss