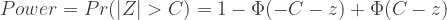背景
近期一篇发表在BMJ Medicine的有关PRS在疾病筛查,预测以及风险分层的文章引发了激烈讨论。
文章:Hingorani, A. D., Gratton, J., Finan, C., Schmidt, A. F., Patel, R., Sofat, R., … & Wald, N. J. (2023). Performance of polygenic risk scores in screening, prediction, and risk stratification: secondary analysis of data in the Polygenic Score Catalog. BMJ medicine, 2(1).
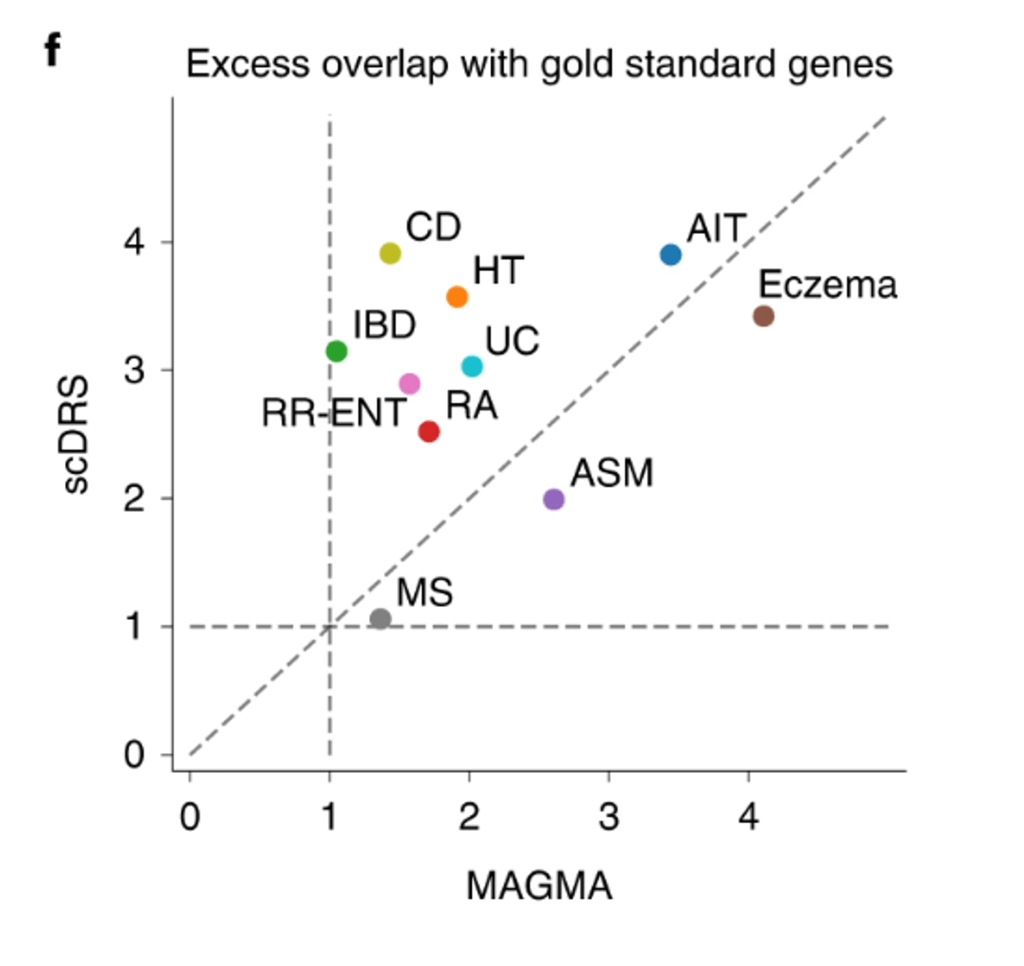
Hingorani等人的文章主要研究对象为PGS Catalog中已发表PGS的性能指标估计值(hazard ratio, odds ratio等), 并将其转化为临床检验常用的指标 DR5 (detection rate for a 5% false positive rate)进行二次分析。这篇文章得出了负面的结论,PRS在疾病筛查,预测以及风险分层等方面表现较差,对PRS的强调与其在健康体系中的实际效果不成正比。
该文章发表于2023年10月17日,随即遭到了曼彻斯特大学D Gareth Evans教授的激烈质疑,“他们关于不要使用PRS的论断就跟说你不应该用任何风险因子评估疾病风险一样。 一个有两倍PRS的女性,如果没有乳腺癌家族史,第一次怀孕过早,和过晚的初潮,其风险也只有平均值。与其他风险因素合并使用可以显著降低文中提到的假阳性以及发现有真的高风险的个体。” 同一天谢菲尔德大学的Harry Hill也对此提出了相似的质疑 。几天后塔尔图大学的Padrik等人则举出乳腺癌的例子进行质疑,他们认为评估PRS对临床的潜在影响,需要在各个疾病的特殊临床背景下进行分析。
几周后本文的第一作者Hingorani对上述质疑一一进行了回应,主要举例数据并引用其他研究说明,即使结合其他风险因子,PRS对于筛查性能的提升是很小的,上述几位的质疑对该文章的主要结论没有影响。
上个月月底,Samuel A. Lambert等一众该领域知名学者(包括PGS Catalog的主要贡献者)联名发文反驳该文的观点,强调PRS既不是诊断性检查,也不是单独的风险因子,并举例说明PRS的应用与其成本效益,指出该文有缺陷的建模与不完整的理论前提。同时致信杂志编辑 “他们的结论基于了不完整的前提,没有考虑临床背景。我们认为他们的文章并没有推动PGS潜在临床应用的讨论,并且会误导临床工作者与大众” 。
目前编辑还没有回复,上面的回复原文可以在 https://bmjmedicine.bmj.com/content/2/1/e000554.responses 找到。有兴趣的同学可以看一看原文。
一些个人理解
(主要基于 Lewis, C. M., & Vassos, E. (2020). Polygenic risk scores: from research tools to clinical instruments. Genome medicine, 12(1), 1-11.)
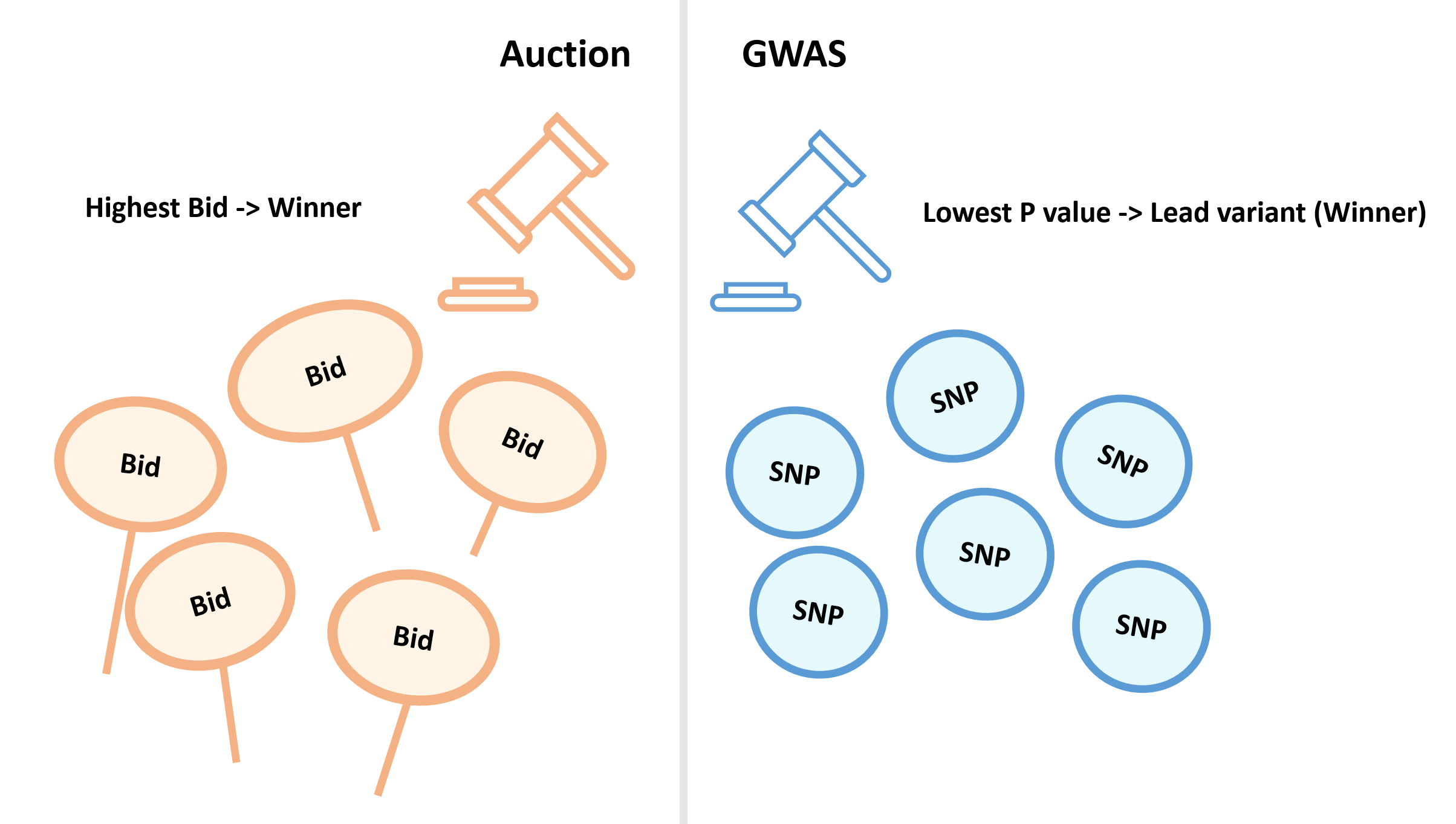
PRS反映的是个体遗传因素相关的疾病风险,对于个体,定义上PRS是独立风险位点的风险等位拷贝数的加权之和。这个模型简单实用,但还有很多不完美之处,例如这个计算方法通常只考虑加性遗传结构,没有考虑可能的基因间相互作用或基因环境相互作用等等,反过来说这些也都是未来PRS方法研究可能的方向。
对于PRS可能的临床应用(疾病预测,风险分层等),应当考虑到PRS的性能会受到多方面影响,例如疾病的多因子性质,遗传信号测量中可能出现的不准确或错误的问题等等。对于复杂疾病,PRS单独使用时性能性能不会太高,解读时,应当认为PRS为传统风险因子预测模型的补充,而非替代。
对于PRS的理解要避免基因决定论的错误印象。如果将PRS单独用于复杂疾病预测就会类似于刻舟求剑,完全忽略了周围不断变化的因素的影响。概念上来讲,个体的遗传易感性(Genetic liability)是固定的,但其所引起的风险却是动态变化的,这个变化依赖于变化的因素例如年龄,环境暴露,家族史,个人病史等等。一个最简单的例子就是,假如一个人有很高的酗酒的遗传风险,但如果他因为一些原因从来没有见过酒,那这个遗传风险自然就无从谈起。
对于PRS的实际应用,还存在诸多需要解决的问题,但相关研究的出发点需要基于对PRS的科学理解,才能避免偏差。
参考
(批判性参考)Hingorani, A. D., Gratton, J., Finan, C., Schmidt, A. F., Patel, R., Sofat, R., … & Wald, N. J. (2023). Performance of polygenic risk scores in screening, prediction, and risk stratification: secondary analysis of data in the Polygenic Score Catalog. BMJ medicine, 2(1). https://bmjmedicine.bmj.com/content/2/1/e000554
Lewis, C. M., & Vassos, E. (2020). Polygenic risk scores: from research tools to clinical instruments. Genome medicine, 12(1), 1-11.





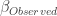 的近似分布为:
的近似分布为: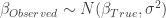
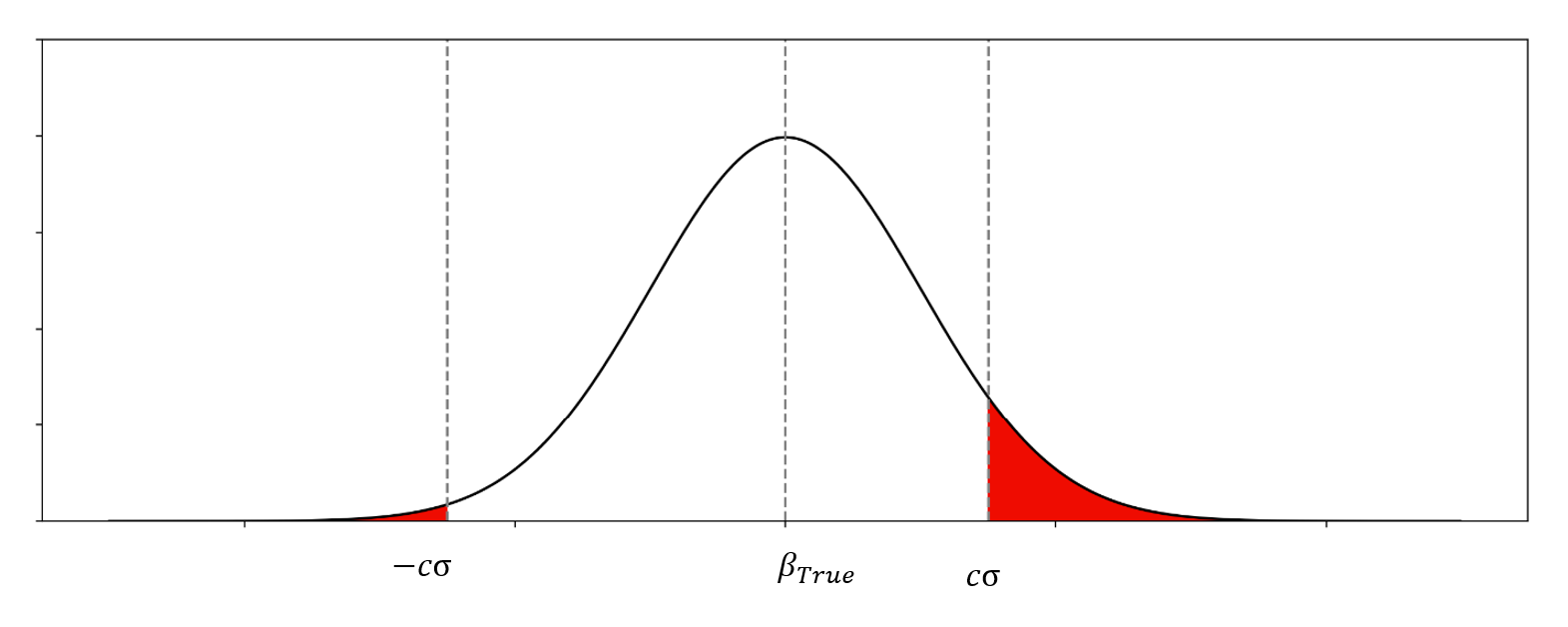

 的一个例子
的一个例子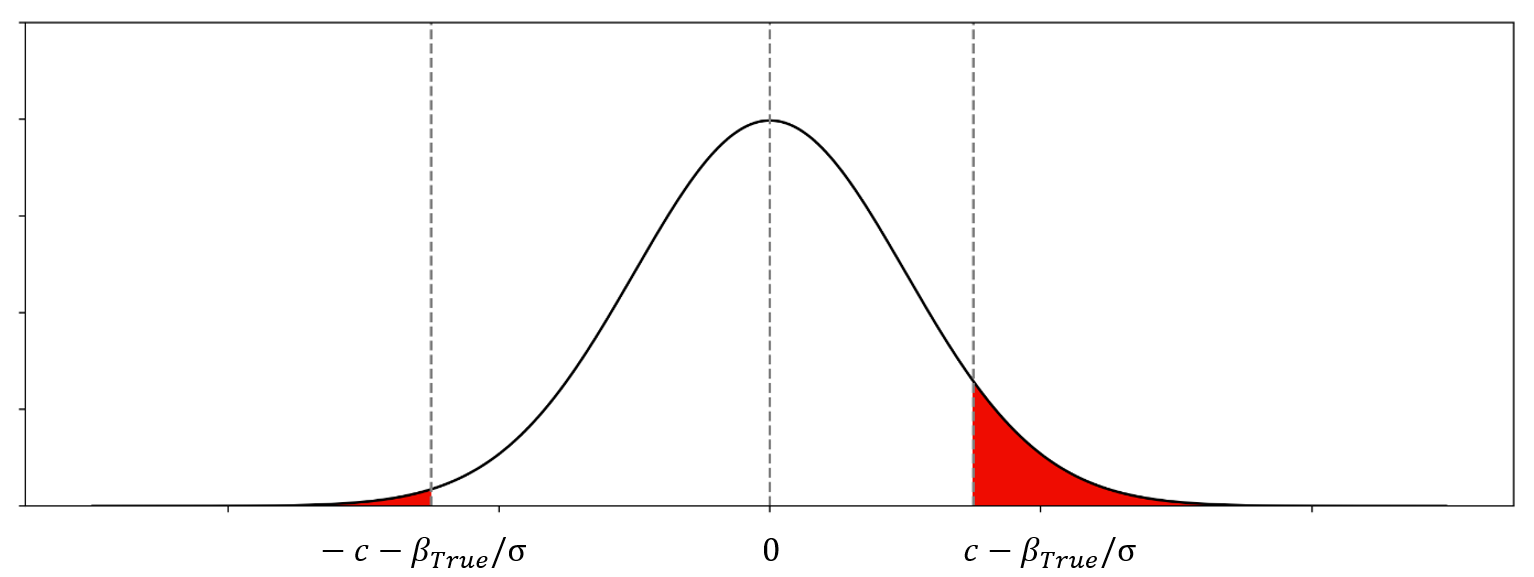

 : 标准正态分布的概率密度函数
: 标准正态分布的概率密度函数 : 标准正态分布的累积分布函数
: 标准正态分布的累积分布函数 
 , SE
, SE 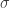 , 以及用于筛选的显著性阈值决定.
, 以及用于筛选的显著性阈值决定. 个样本为AB基因型的精确概率为:
个样本为AB基因型的精确概率为:
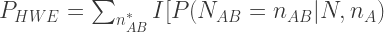
![\geqq P(N_{AB} = n^{*}_{AB} | N, n_A)] \times P(N_{AB} = n^{*}_{AB} | N, n_A)](https://s0.wp.com/latex.php?latex=%5Cgeqq+P%28N_%7BAB%7D+%3D+n%5E%7B%2A%7D_%7BAB%7D+%7C+N%2C+n_A%29%5D+%5Ctimes+P%28N_%7BAB%7D+%3D+n%5E%7B%2A%7D_%7BAB%7D+%7C+N%2C+n_A%29+&bg=ffffff&fg=333333&s=2&c=20201002)
 为一个指示函数. 如果x为真,
为一个指示函数. 如果x为真,  ; 否则,
; 否则,  .
.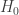 与统计学检验结果(是否拒绝原假设
与统计学检验结果(是否拒绝原假设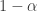

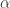
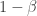

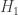 之间差异的程度。
之间差异的程度。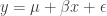



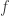 : 该变异的等位频率(allele frequency)
: 该变异的等位频率(allele frequency)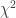 分布的非中心参数NCP则为
分布的非中心参数NCP则为
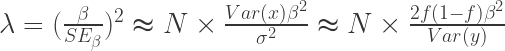
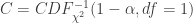
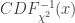 :
: 
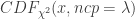 : 非中心参数NCP为
: 非中心参数NCP为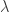 的
的 : 在病例中风险等位的频率 Risk allele frequency in cases
: 在病例中风险等位的频率 Risk allele frequency in cases : 病例的样本量 Number of cases. The total allele count for cases is then
: 病例的样本量 Number of cases. The total allele count for cases is then  .
.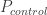 : 在对照中风险等位的频率 Risk allele frequency in controls
: 在对照中风险等位的频率 Risk allele frequency in controls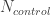 : 对照的样本量 Number of control. The total allele count for control is then
: 对照的样本量 Number of control. The total allele count for control is then  .
. , 即风险等位的频率在病例中与对照中是一样的。
, 即风险等位的频率在病例中与对照中是一样的。