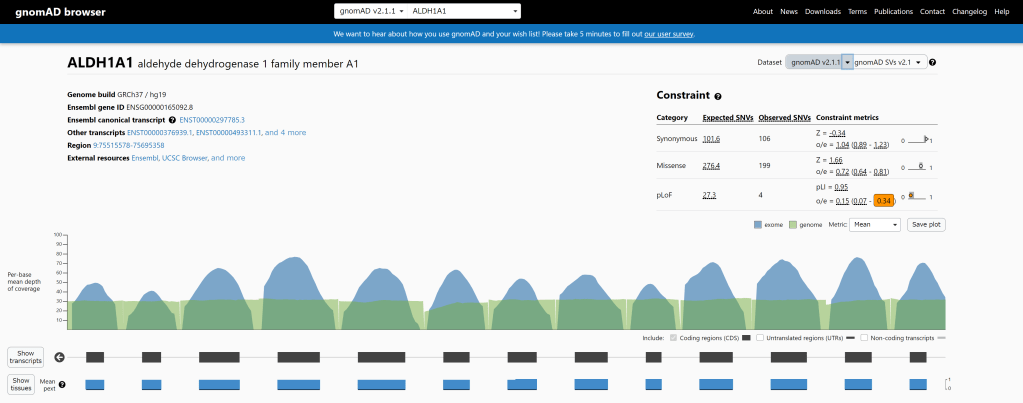
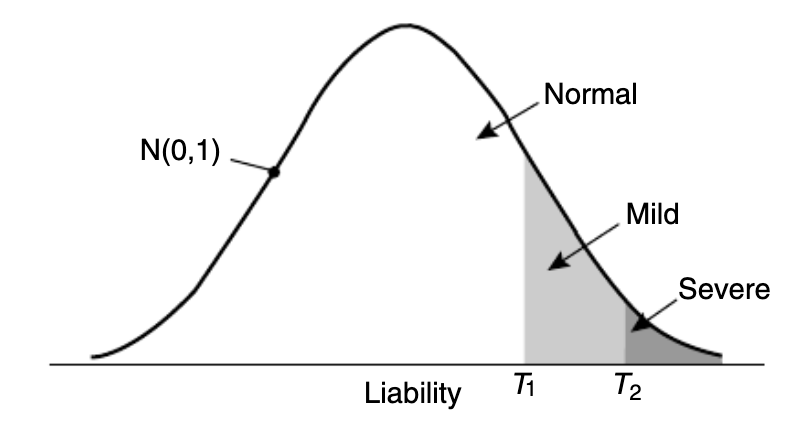
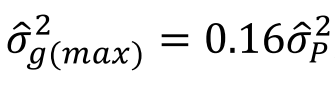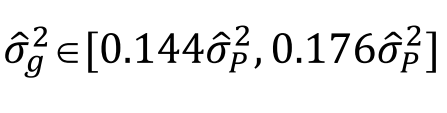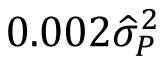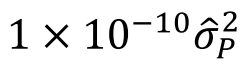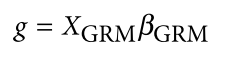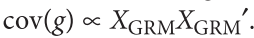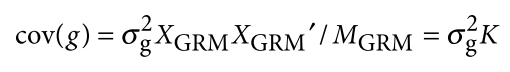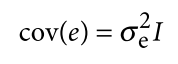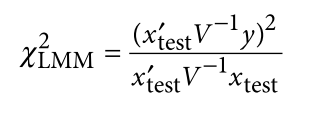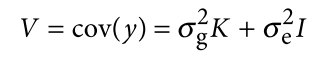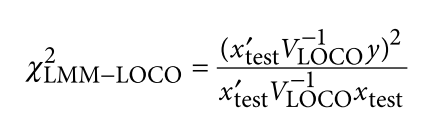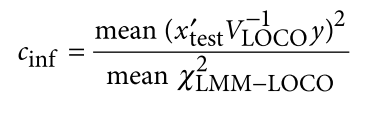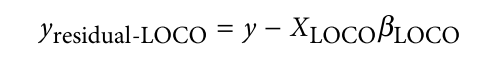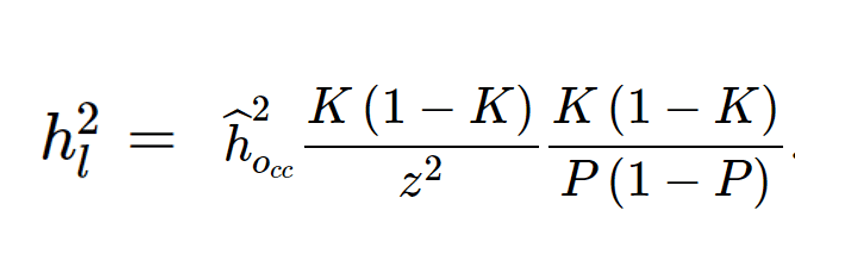遗传学中关于亲缘关系的常见的几个系数辨析:
血缘系数 coefficient of kinship / kinship coefficient,有些地方称为近亲系数,有时也称共祖系数 coefficient of coancestry, (目前缺少权威的中文翻译),是对个体间血缘关系的直接衡量,定义为从两个个体随机抽取一个同源等位基因,所抽取的两个等位基因是血缘同源(IBD)的概率(即这两个等位基因相同,且来自同一个祖先)。
常用的用来衡量亲缘关系的,且容易混淆的系数还有如下两个,近交系数与血缘系数。
近交系数 coefficient of inbreeding ,由怀特( Wright, Sewall )最早定义,是指 一个个体的某个基因座上的两个等位基因为血缘同源 (IBD) 的概率,衡量的是这个个体父母间亲缘程度的大小,反映的是近亲交配的程度。有时也称为固定系数 fixation index ,通常用 F 表示。
亲缘系数 coefficient of relationship / coefficient of relatedness ,该系数也由怀特定义,衍生自他对近交系数的定义, 是指有共同祖先的两个个体间,基因型一致的概率,通常用 r 来表示。数值上是 血缘系数 的两倍。
对于常见亲缘关系这三个系数的理论值如下图所示:
| 与个体的关系 | 血缘系数 | 近交系数 F | 亲缘系数 r |
| 自己,同卵双胞胎 | 1/2 | 1/2 | 1 |
| 亲兄弟姐妹 | 1/4 | 1/4 | 1/2 |
| 母父,儿女* | 1/4 | 1/4 | 1/2 |
| 祖父母,孙子孙女 | 1/8 | 1/8 | 1/4 |
| 舅舅,舅妈,侄女,侄子 | 1/8 | 1/8 | 1/4 |
| 表兄弟 | 1/16 | 1/16 | 1/8 |
| 同父异母或同母异父的兄弟姐妹 | 1/8 | 1/8 | 1/4 |
*注意,虽然近交系数与血缘系数值相等,当他们的概念并不相同。
血缘系数是指两个个体间的血缘关系,如 ( 母父,儿女 )这一项,血缘系数为1/4的意思是从一个个体与其亲生父母或孩子个随机抽取一个同源等位基因,这两个等位基因相同,且来自同一个祖先 的概率是1/4.
而近交系数在这里的值为虽然也为1/4,但其表达的意思是如果这个个体与其 亲生父母或孩子近交后,子代的某个基因座上的两个等位基因为血缘同源的概率 为1/4.
参考:
Wright, Sewall (1922). “Coefficients of inbreeding and relationship”. American Naturalist. 56 (645): 330–338. doi:10.1086/279872.





 是某个等位基因在整个群体里的频率,
是某个等位基因在整个群体里的频率,  是等位基因在不同亚群体之间的被群体大小加权后的频率的方差(组间方差),
是等位基因在不同亚群体之间的被群体大小加权后的频率的方差(组间方差), 是整个群体的等位基因频率的方差。那么Fst可以被定义为:
是整个群体的等位基因频率的方差。那么Fst可以被定义为:


 则是 给定两个来自总体的个体,这两个个体血缘同源(IBD)的概率。
则是 给定两个来自总体的个体,这两个个体血缘同源(IBD)的概率。 
 与
与 分别代表两个不同亚群或相同亚群的个体之间,成对等位基因之间不同的平均值(average number of pairwise differences)。
分别代表两个不同亚群或相同亚群的个体之间,成对等位基因之间不同的平均值(average number of pairwise differences)。